 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-1457: A test-bed for optimizing high-resolution single particle reconst... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-1457 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | A test-bed for optimizing high-resolution single particle reconstructions. | |||||||||
 Map data Map data | This is a single particle reconstruction of GroEL | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / negative staining / Resolution: 5.4 Å | |||||||||
 Authors Authors | Stagg SM / Lander GC / Quispe J / Voss NR / Cheng A / Bradlow H / Bradlow S / Carragher B / Potter CS | |||||||||
 Citation Citation | Journal: J Struct Biol / Year: 2008 Title: A test-bed for optimizing high-resolution single particle reconstructions. Authors: Scott M Stagg / Gabriel C Lander / Joel Quispe / Neil R Voss / Anchi Cheng / Henry Bradlow / Steven Bradlow / Bridget Carragher / Clinton S Potter /  Abstract: It is becoming routine for cryoEM single particle reconstructions to result in 3D electron density maps with resolutions of approximately 10A, but maps with resolutions of 5A or better are still ...It is becoming routine for cryoEM single particle reconstructions to result in 3D electron density maps with resolutions of approximately 10A, but maps with resolutions of 5A or better are still celebrated events. The electron microscope has a resolving power to better than 2A, and thus should not be a limiting factor; instead the practical limitations in resolution most likely arise from a combination of specimen preparation methods, data collection parameters, and data analysis procedures. With the aid of a highly automated system for acquiring images, coupled to a relational database to keep track of all processing parameters, we have taken a systematic approach to optimizing parameters affecting the resolution of single particle reconstructions. Using GroEL as a test-bed, we performed a series of 3D reconstructions where we systematically varied the number of particles used in computing the map, the accelerating voltage of the microscope, and the electron dose used to acquire the images. We also investigated methods for excluding unacceptable or "bad" particles from contributing to the final 3D map. Using relatively standard instrumentation (Tecnai F20, 4K x 4K CCD, side entry cold stage) and a completely automated approach, these approaches resulted in a map with a nominal resolution of 5.4A (FSC(0.5)) in which secondary structure is clearly discernable and the handedness of some of the alpha-helices in the GroEL structure can be determined. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_1457.map.gz | 5.2 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-1457-v30.xmlemd-1457.xml | 9.7 KB 9.7 KB | Display Display | EMDB header |
| Images |  1457.gif 1457.gif | 59.3 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-1457ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1457 http://ftp.pdbj.org/pub/emdb/structures/EMD-1457ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1457 | HTTPS FTP |
-Related structure data











-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_1457.map.gz / Format: CCP4 / Size: 14.1 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | This is a single particle reconstruction of GroEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
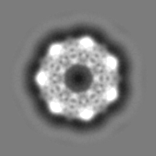
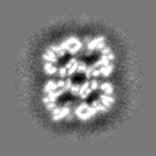
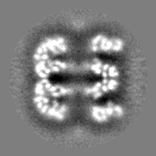
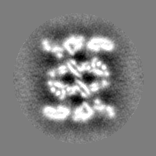
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.63 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)
















-Supplemental data
- Sample components
Sample components
-Entire : GroEL
| Entire | Name: GroEL |
|---|---|
| Components |
|
-Supramolecule #1000: GroEL
| Supramolecule | Name: GroEL / type: sample / ID: 1000 / Oligomeric state: D7 14-mer / Number unique components: 1 |
|---|---|
| Molecular weight | Experimental: 800 KDa / Theoretical: 800 KDa |
-Macromolecule #1: GroEL
| Macromolecule | Name: GroEL / type: protein_or_peptide / ID: 1 / Name.synonym: GroEL / Number of copies: 14 / Oligomeric state: homotetradecamer / Recombinant expression: Yes |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Experimental: 800 KDa / Theoretical: 800 KDa |
-Experimental details
-Structure determination
| Method | negative staining, cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 4 mg/mL |
|---|---|
| Buffer | pH: 7.5 / Details: 100mM Hepes, 10mM Mg(OAc)2, 10mM KOAc, 2mM DTT |
| Staining | Type: NEGATIVE / Details: not stained |
| Grid | Details: Protochips C-flat grid: holey carbon with 2um holes and 2um spacing 400 mesh copper grid |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 93 K / Instrument: OTHER Details: Vitrification instrument: Vitrobot. Grid plasma cleaned for 20s with Fischione 1020 plasma cleaner using 75% Argon 25% Oxygen mix. Method: Temperature of chamber was 4 degrees C. 0 seconds drain time. Single blot. 0 mm offset. 4 ul sample applied to grid. Blot for 3.5 seconds before plunging. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI F20 |
|---|---|
| Temperature | Average: 102 K |
| Alignment procedure | Legacy - Astigmatism: objective lens astigmatism was corrected automatically using Leginon |
| Date | Jul 12, 2006 |
| Image recording | Category: CCD / Film or detector model: GATAN ULTRASCAN 4000 (4k x 4k) / Average electron dose: 13 e/Å2 |
| Tilt angle min | 0 |
| Tilt angle max | 0 |
| Electron beam | Acceleration voltage: 120 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 100000 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2 mm / Nominal defocus max: 3.0 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 100000 |
| Sample stage | Specimen holder: Side entry liquid nitrogen-cooled cryo specimen holder Specimen holder model: GATAN LIQUID NITROGEN |
| Experimental equipment |  Model: Tecnai F20 / Image courtesy: FEI Company |
-Image processing
| CTF correction | Details: Phase correction for each particle. |
|---|---|
| Final reconstruction | Applied symmetry - Point group: D7 (2x7 fold dihedral) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 5.4 Å / Resolution method: FSC 0.5 CUT-OFF / Software - Name: EMAN / Number images used: 55351 |
| Final two d classification | Number classes: 1294 |
