 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-1016: Difference imaging of adenovirus: bridging the resolution gap bet... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-1016 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Difference imaging of adenovirus: bridging the resolution gap between X-ray crystallography and electron microscopy. | |||||||||
 Map data Map data | Adeno-virus type 2. | |||||||||
 Sample Sample |
| |||||||||
| Biological species |   Human adenovirus 2 Human adenovirus 2 | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 25.0 Å | |||||||||
 Authors Authors | Stewart PL / Fuller SD / Burnett RM | |||||||||
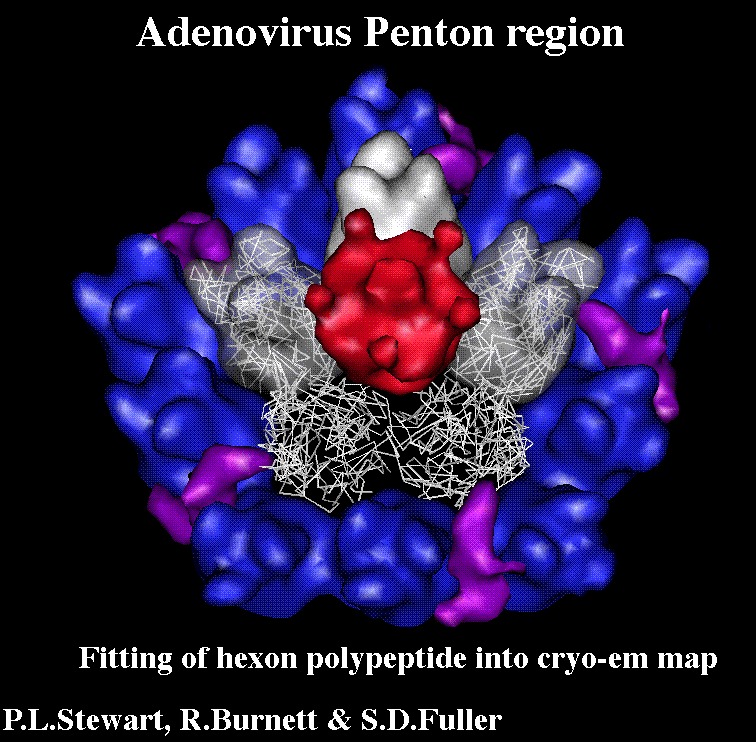
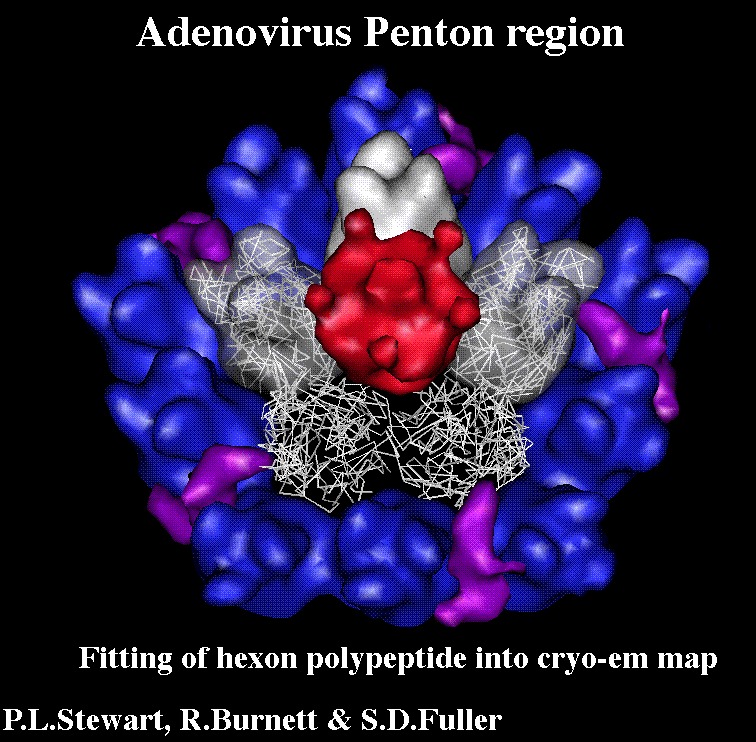
 Citation Citation | Journal: EMBO J / Year: 1993 Title: Difference imaging of adenovirus: bridging the resolution gap between X-ray crystallography and electron microscopy. Authors: P L Stewart / S D Fuller / R M Burnett /  Abstract: While X-ray crystallography provides atomic resolution structures of proteins and small viruses, electron microscopy provides complementary structural information on the organization of larger ...While X-ray crystallography provides atomic resolution structures of proteins and small viruses, electron microscopy provides complementary structural information on the organization of larger assemblies at lower resolution. A novel combination of these two techniques has bridged this resolution gap and revealed the various structural components forming the capsid of human type 2 adenovirus. An image reconstruction of the intact virus, derived from cryo-electron micrographs, was deconvolved with an approximate contrast transfer function to mitigate microscope distortions. A model capsid was calculated from 240 copies of the crystallographic structure of the major capsid protein and filtered to the correct resolution. Subtraction of the calculated capsid from the corrected reconstruction gave a three-dimensional difference map revealing the minor proteins that stabilize the virion. Elongated density penetrating the hexon capsid at the facet edges was ascribed to polypeptide IIIa, a component required for virion assembly. Density on the inner surface of the capsid, connecting the ring of peripentonal hexons, was assigned as polypeptide VI, a component that binds DNA. Identification of the regions of hexon that contact the penton base suggests a structural mechanism for previously proposed events during cell entry. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
 UCSF Chimera
UCSF Chimera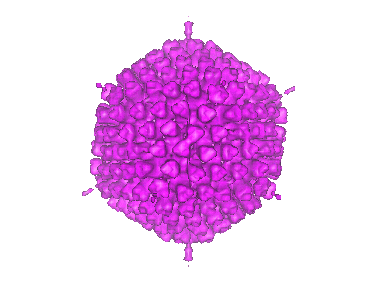
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_1016.map.gz | 2.1 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-1016-v30.xmlemd-1016.xml | 15.8 KB 15.8 KB | Display Display | EMDB header |
| Images |  1016.gifemd_1016.tifemd_emd_1016.tif 1016.gifemd_1016.tifemd_emd_1016.tif | 28.8 KB 108 KB 108 KB | ||
| Others | emd_1016_additional_1.map.gzemd_1016_additional_2.map.gzemd_1016_additional_3.map.gz | 23.1 KB 22.6 KB 24.2 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-1016ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1016 http://ftp.pdbj.org/pub/emdb/structures/EMD-1016ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1016 | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|









-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_1016.map.gz / Format: CCP4 / Size: 7.8 MB / Type: IMAGE STORED AS SIGNED INTEGER (2 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Adeno-virus type 2. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 7.3 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Supplemental map: emd 1016 additional 1.map
| File | emd_1016_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Supplemental map: emd 1016 additional 2.map
| File | emd_1016_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Supplemental map: emd 1016 additional 3.map
| File | emd_1016_additional_3.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : adenovirus type 2
| Entire | Name: adenovirus type 2 |
|---|---|
| Components |
|
-Supramolecule #1000: adenovirus type 2
| Supramolecule | Name: adenovirus type 2 / type: sample / ID: 1000 / Oligomeric state: T=25 particle / Number unique components: 1 |
|---|---|
| Molecular weight | Theoretical: 150 MDa |
-Supramolecule #1: Human adenovirus 2
| Supramolecule | Name: Human adenovirus 2 / type: virus / ID: 1 / NCBI-ID: 10515 / Sci species name: Human adenovirus 2 / Virus type: VIRION / Virus isolate: STRAIN / Virus enveloped: No / Virus empty: No |
|---|---|
| Host (natural) | Organism:  Homo sapiens (human) / synonym: VERTEBRATES Homo sapiens (human) / synonym: VERTEBRATES |
| Virus shell | Shell ID: 1 / Name: fibres (5f positions) 1090A / T number (triangulation number): 25 |
| Virus shell | Shell ID: 2 / Name: capsid (along 2f) 884A / T number (triangulation number): 1 |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 10 mg/mL |
|---|---|
| Buffer | pH: 7.4 Details: Sample stored in CsCl was diluted in Tris (10mM) NaCl (100 mM) pH 7.4 |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 60 % / Chamber temperature: 23 K / Instrument: HOMEMADE PLUNGER Details: Vitrification instrument: EMBL plunger. plunging at ambient temperature and humidity Method: Blot for 2 sec |
- Electron microscopy
Electron microscopy
| Microscope | FEI/PHILIPS EM400 |
|---|---|
| Temperature | Average: 105 K |
| Details | Details of the microscopy are in Stewart, P. L., Burnett, R. M., Cyrklaff, M. and Fuller, S. D. (1991). Image reconstruction reveals the complex molecular organization of adenovirus. Cell 67, 145-54. Images were taken in triples at 1, 2.6 and 8.8 microns underfocus. Some images were taken at 12 degrees tilt to compensate for the preference for the virus to orient with the fibres away from the water layer. For image reconstruction see: Stewart, P. L., Burnett, R. M.,Cyrklaff, M. and Fuller, S. D. (1991). Image reconstruction reveals the complex molecular organization of adenovirus. Cell 67, 145-54. For reconstruction details see: P.L. Stewart, S.D. Fuller, R. M. Burnett, (1993). Difference imaging of adenovirus: bridging the resolution gap between X- ray crystallography and electron microscopy EMBO J.Stewart PL, Burnett RM. Adenovirus structure by X-ray crystallography and electron microscopy. Curr Top Microbiol Immunol. 1995, 199 (1):25-38. The PDB code for the Ad2 hexon model is 1DHX, and the reference Athappilly, F. K., Murali, R., Rux, J. J., Cai, Z., Burnett, R. M.: The refined crystal structure of hexon, the major coat protein of adenovirus type 2, at 2.9 A resolution. J Mol Biol 242 pp. 430 (1994) |
| Date | Jan 1, 1990 |
| Image recording | Category: FILM / Film or detector model: KODAK SO-163 FILM / Digitization - Scanner: OPTRONICS / Digitization - Sampling interval: 25 µm / Number real images: 50 / Average electron dose: 8 e/Å2 / Od range: 1 / Bits/pixel: 8 |
| Electron beam | Acceleration voltage: 80 kV / Electron source: TUNGSTEN HAIRPIN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2 mm / Nominal defocus max: 8.8 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 34000 |
| Sample stage | Specimen holder: eucentric - Philips EM400 / Specimen holder model: GATAN LIQUID NITROGEN / Tilt angle max: 12 |
-Image processing
| Details | Purified by centrifugation in CsCl |
|---|---|
| CTF correction | Details: initially different defocuses were combined by summing in resolution bands |
| Final reconstruction | Applied symmetry - Point group: I (icosahedral) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 25.0 Å / Resolution method: OTHER / Software - Name: EMBL-ICOS / Number images used: 29 |
| Final angle assignment | Details: sufficient to give maximum inverse eigenvalue of 0.1 |
-Atomic model buiding 1
| Software | Name: maximizing cross correlation |
|---|---|
| Details | Protocol: rigid body |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Overall B value: 3000 / Target criteria: max cross correlation |
