 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-0325 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
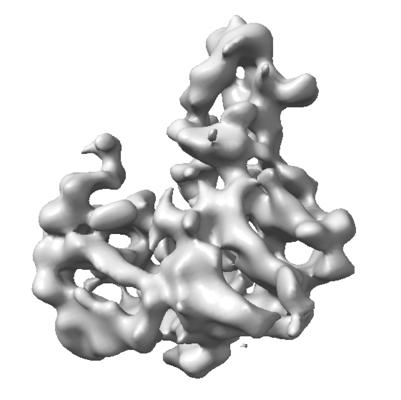
| Title | Cryo-EM map of the Ysh1-Mpe1 nuclease complex | ||||||||||||
 Map data Map data | Cryo-EM map of the Ysh1-Mpe1 complex | ||||||||||||
 Sample Sample |
| ||||||||||||
| Biological species |  | ||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 6.0 Å | ||||||||||||
 Authors Authors | Hill CH / Boreikaite V / Kumar A / Casanal A / Kubik P / Degliesposti G / Maslen S / Mariani A / von Loeffelholz O / Girbig M ...Hill CH / Boreikaite V / Kumar A / Casanal A / Kubik P / Degliesposti G / Maslen S / Mariani A / von Loeffelholz O / Girbig M / Skehel M / Passmore LA | ||||||||||||
| Funding support |  United Kingdom, 3 items United Kingdom, 3 items
| ||||||||||||
 Citation Citation | Journal: Mol Cell / Year: 2019 Title: Activation of the Endonuclease that Defines mRNA 3' Ends Requires Incorporation into an 8-Subunit Core Cleavage and Polyadenylation Factor Complex. Authors: Chris H Hill / Vytautė Boreikaitė / Ananthanarayanan Kumar / Ana Casañal / Peter Kubík / Gianluca Degliesposti / Sarah Maslen / Angelica Mariani / Ottilie von Loeffelholz / Mathias ...Authors: Chris H Hill / Vytautė Boreikaitė / Ananthanarayanan Kumar / Ana Casañal / Peter Kubík / Gianluca Degliesposti / Sarah Maslen / Angelica Mariani / Ottilie von Loeffelholz / Mathias Girbig / Mark Skehel / Lori A Passmore /  Abstract: Cleavage and polyadenylation factor (CPF/CPSF) is a multi-protein complex essential for formation of eukaryotic mRNA 3' ends. CPF cleaves pre-mRNAs at a specific site and adds a poly(A) tail. The ...Cleavage and polyadenylation factor (CPF/CPSF) is a multi-protein complex essential for formation of eukaryotic mRNA 3' ends. CPF cleaves pre-mRNAs at a specific site and adds a poly(A) tail. The cleavage reaction defines the 3' end of the mature mRNA, and thus the activity of the endonuclease is highly regulated. Here, we show that reconstitution of specific pre-mRNA cleavage with recombinant yeast proteins requires incorporation of the Ysh1 endonuclease into an eight-subunit "CPF" complex. Cleavage also requires the accessory cleavage factors IA and IB, which bind substrate pre-mRNAs and CPF, likely facilitating assembly of an active complex. Using X-ray crystallography, electron microscopy, and mass spectrometry, we determine the structure of Ysh1 bound to Mpe1 and the arrangement of subunits within CPF. Together, our data suggest that the active mRNA 3' end processing machinery is a dynamic assembly that is licensed to cleave only when all protein factors come together at the polyadenylation site. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_0325.map.gz | 1.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-0325-v30.xmlemd-0325.xml | 21.6 KB 21.6 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_0325_fsc.xml | 4.7 KB | Display | FSC data file |
| Images |  emd_0325.png emd_0325.png | 56.5 KB | ||
| Masks | emd_0325_msk_1.map | 8 MB | Mask map | |
| Others | emd_0325_half_map_1.map.gzemd_0325_half_map_2.map.gz | 6 MB 6 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-0325ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0325 http://ftp.pdbj.org/pub/emdb/structures/EMD-0325ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0325 | HTTPS FTP |
-Related structure data











-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_0325.map.gz / Format: CCP4 / Size: 8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Cryo-EM map of the Ysh1-Mpe1 complex | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.09 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_0325_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
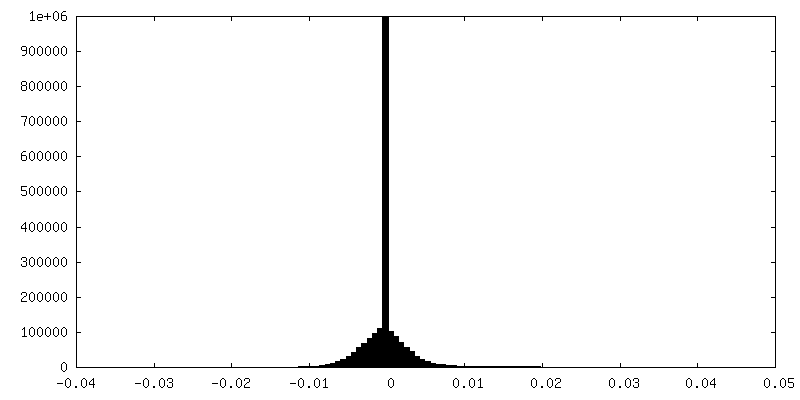
| Density Histograms |






-Half map: half-map 1
| File | emd_0325_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half-map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |



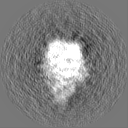
-Half map: half-map 2
| File | emd_0325_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half-map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |



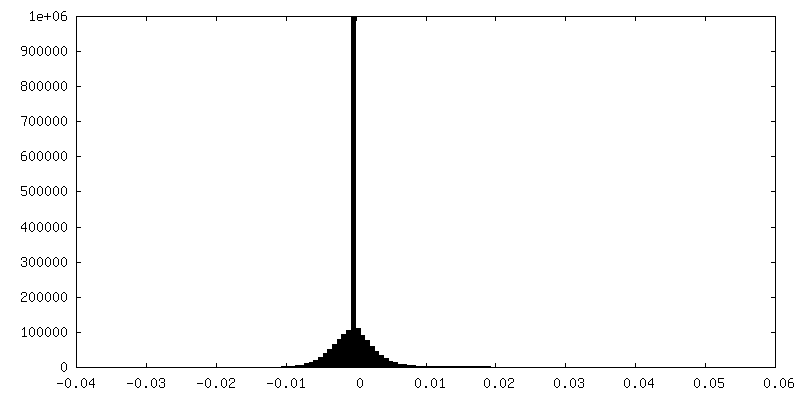
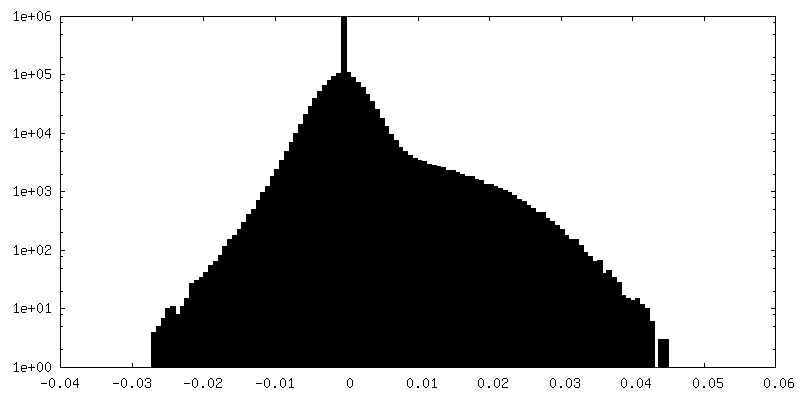
- Sample components
Sample components
-Entire : Ysh1-Mpe1 complex Highly anisotropic due to severe preferred orie...
| Entire | Name: Ysh1-Mpe1 complex Highly anisotropic due to severe preferred orientation. |
|---|---|
| Components |
|
-Supramolecule #1: Ysh1-Mpe1 complex Highly anisotropic due to severe preferred orie...
| Supramolecule | Name: Ysh1-Mpe1 complex Highly anisotropic due to severe preferred orientation. type: complex / ID: 1 / Parent: 0 / Macromolecule list: all Details: The trimeric Ysh1-Mpe1-Yjr141w complex was analysed, however several regions are flexible/disordered. This map corresponds to the Ysh1 N-terminal nuclease domain in complex with the Mpe1 N- ...Details: The trimeric Ysh1-Mpe1-Yjr141w complex was analysed, however several regions are flexible/disordered. This map corresponds to the Ysh1 N-terminal nuclease domain in complex with the Mpe1 N-terminal ubiquitin-like domain. These are the only ordered, globular densities present in the above sample. |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism:  unidentified baculovirus / Recombinant cell: Sf9 unidentified baculovirus / Recombinant cell: Sf9 |
| Molecular weight | Theoretical: 57 KDa |
-Macromolecule #1: Ysh1
| Macromolecule | Name: Ysh1 / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism: unidentified baculovirus |
| Sequence | String: MERTNTTTFK FFSLGGSNEV GRSCHILQYK GKTVMLDAGI HPAYQGLASL PFYDEFDLSK VDILLISHF HLDHAASLPY VMQRTNFQGR VFMTHPTKAI YRWLLRDFVR VTSIGSSSSS M GTKDEGLF SDEDLVDSFD KIETVDYHST VDVNGIKFTA FHAGHVLGAA ...String: MERTNTTTFK FFSLGGSNEV GRSCHILQYK GKTVMLDAGI HPAYQGLASL PFYDEFDLSK VDILLISHF HLDHAASLPY VMQRTNFQGR VFMTHPTKAI YRWLLRDFVR VTSIGSSSSS M GTKDEGLF SDEDLVDSFD KIETVDYHST VDVNGIKFTA FHAGHVLGAA MFQIEIAGLR VL FTGDYSR EVDRHLNSAE VPPLSSNVLI VESTFGTATH EPRLNRERKL TQLIHSTVMR GGR VLLPVF ALGRAQEIML ILDEYWSQHA DELGGGQVPI FYASNLAKKC MSVFQTYVNM MNDD IRKKF RDSQTNPFIF KNISYLRNLE DFQDFGPSVM LASPGMLQSG LSRDLLERWC PEDKN LVLI TGYSIEGTMA KFIMLEPDTI PSINNPEITI PRRCQVEEIS FAAHVDFQEN LEFIEK ISA PNIILVHGEA NPMGRLKSAL LSNFASLKGT DNEVHVFNPR NCVEVDLEFQ GVKVAKA VG NIVNEIYKEE NVEIKEEIAA KIEPIKEENE DNLDSQAEKG LVDEEEHKDI VVSGILVS D DKNFELDFLS LSDLREHHPD LSTTILRERQ SVRVNCKKEL IYWHILQMFG EAEVLQDDD RVTNQEPKVK EESKDNLTNT GKLILQIMGD IKLTIVNTLA VVEWTQDLMN DTVADSIIAI LMNVDSAPA SVKLSSHSCD DHDHNNVQSN AQGKIDEVER VKQISRLFKE QFGDCFTLFL N KDEYASNK EETITGVVTI GKSTAKIDFN NMKILECNSN PLKGRVESLL NIGGNLVTPL C |
-Macromolecule #2: Mpe1
| Macromolecule | Name: Mpe1 / type: protein_or_peptide / ID: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism: unidentified baculovirus |
| Sequence | String: MSSTIFYRFK SQRNTSRILF DGTGLTVFDL KREIIQENKL GDGTDFQLKI YNPDTEEEYD DDAFVIPRS TSVIVKRSPA IKSFSVHSRL KGNVGAAALG NATRYVTGRP RVLQKRQHTA T TTANVSGT TEEERIASMF ATQENQWEQT QEEMSAATPV FFKSQTNKNS ...String: MSSTIFYRFK SQRNTSRILF DGTGLTVFDL KREIIQENKL GDGTDFQLKI YNPDTEEEYD DDAFVIPRS TSVIVKRSPA IKSFSVHSRL KGNVGAAALG NATRYVTGRP RVLQKRQHTA T TTANVSGT TEEERIASMF ATQENQWEQT QEEMSAATPV FFKSQTNKNS AQENEGPPPP GY MCYRCGG RDHWIKNCPT NSDPNFEGKR IRRTTGIPKK FLKSIEIDPE TMTPEEMAQR KIM ITDEGK FVVQVEDKQS WEDYQRKREN RQIDGDETIW RKGHFKDLPD DLKCPLTGGL LRQP VKTSK CCNIDFSKEA LENALVESDF VCPNCETRDI LLDSLVPDQD KEKEVETFLK KQEEL HGSS KDGNQPETKK MKLMDPTGTA GLNNNTSLPT SVNNGGTPVP PVPLPFGIPP FPMFPM PFM PPTATITNPH QADASPKK |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.9 Component:
| ||||||
|---|---|---|---|---|---|---|---|
| Grid | Model: Quantifoil, UltrAuFoil / Material: GOLD / Mesh: 300 / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Atmosphere: OTHER | ||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV / Details: blotted for 10 s. | ||||||
| Details | 350 nM complex was used. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Phase plate: VOLTA PHASE PLATE / Spherical aberration corrector: hexapole Cs-corrector / Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Digitization - Frames/image: 1-42 / Number grids imaged: 1 / Number real images: 994 / Average exposure time: 9.0 sec. / Average electron dose: 45.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
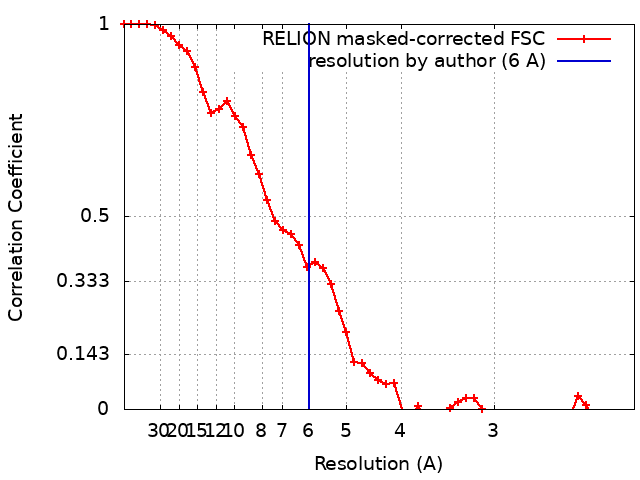
-Atomic model buiding 1
| Initial model | PDB ID: |
|---|---|
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
