 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8167 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of S. cerevesiae mApe1 dodecamer | |||||||||
 Map data Map data | None | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | dodecamer / aminopeptidase / vacuole / cvt / Hydrolase | |||||||||
| Function / homology |  Function and homology information Function and homology informationaminopeptidase I / Cvt complex / cytoplasm to vacuole targeting by the Cvt pathway / fungal-type vacuole / metalloaminopeptidase activity / proteolysis / zinc ion binding / identical protein binding / cytoplasm Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / negative staining / Resolution: 24.0 Å | |||||||||
 Authors Authors | Sachse C / Bertipaglia C | |||||||||
 Citation Citation | Journal: EMBO Rep / Year: 2016 Title: Higher-order assemblies of oligomeric cargo receptor complexes form the membrane scaffold of the Cvt vesicle. Authors: Chiara Bertipaglia / Sarah Schneider / Arjen J Jakobi / Abul K Tarafder / Yury S Bykov / Andrea Picco / Wanda Kukulski / Jan Kosinski / Wim Jh Hagen / Arvind C Ravichandran / Matthias ...Authors: Chiara Bertipaglia / Sarah Schneider / Arjen J Jakobi / Abul K Tarafder / Yury S Bykov / Andrea Picco / Wanda Kukulski / Jan Kosinski / Wim Jh Hagen / Arvind C Ravichandran / Matthias Wilmanns / Marko Kaksonen / John Ag Briggs / Carsten Sachse /  Abstract: Selective autophagy is the mechanism by which large cargos are specifically sequestered for degradation. The structural details of cargo and receptor assembly giving rise to autophagic vesicles ...Selective autophagy is the mechanism by which large cargos are specifically sequestered for degradation. The structural details of cargo and receptor assembly giving rise to autophagic vesicles remain to be elucidated. We utilize the yeast cytoplasm-to-vacuole targeting (Cvt) pathway, a prototype of selective autophagy, together with a multi-scale analysis approach to study the molecular structure of Cvt vesicles. We report the oligomeric nature of the major Cvt cargo Ape1 with a combined 2.8 Å X-ray and negative stain EM structure, as well as the secondary cargo Ams1 with a 6.3 Å cryo-EM structure. We show that the major dodecameric cargo prApe1 exhibits a tendency to form higher-order chain structures that are broken upon interaction with the receptor Atg19 in vitro The stoichiometry of these cargo-receptor complexes is key to maintaining the size of the Cvt aggregate in vivo Using correlative light and electron microscopy, we further visualize key stages of Cvt vesicle biogenesis. Our findings suggest that Atg19 interaction limits Ape1 aggregate size while serving as a vehicle for vacuolar delivery of tetrameric Ams1. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
 UCSF Chimera
UCSF Chimera
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8167.map.gz | 1.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8167-v30.xmlemd-8167.xml | 12 KB 12 KB | Display Display | EMDB header |
| Images |  emd_8167.png emd_8167.png | 175.1 KB | ||
| Filedesc metadata | emd-8167.cif.gz | 6 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8167ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8167 http://ftp.pdbj.org/pub/emdb/structures/EMD-8167ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8167 | HTTPS FTP |
-Related structure data
| Related structure data |  5jm9MC  8166C  5jm0C  5jm6C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_8167.map.gz / Format: CCP4 / Size: 7.5 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | None | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.9 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
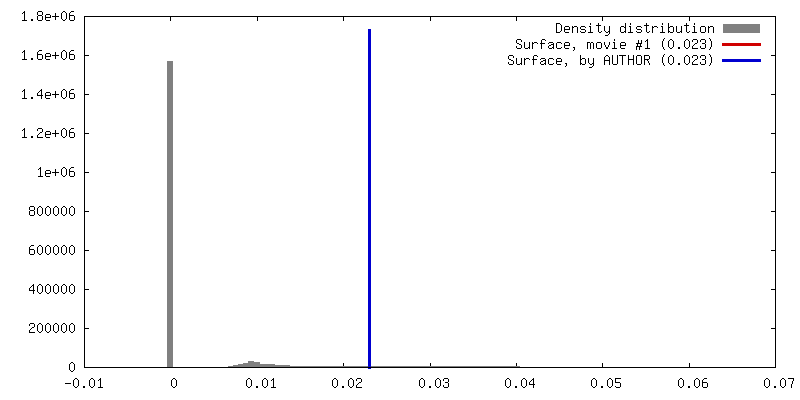
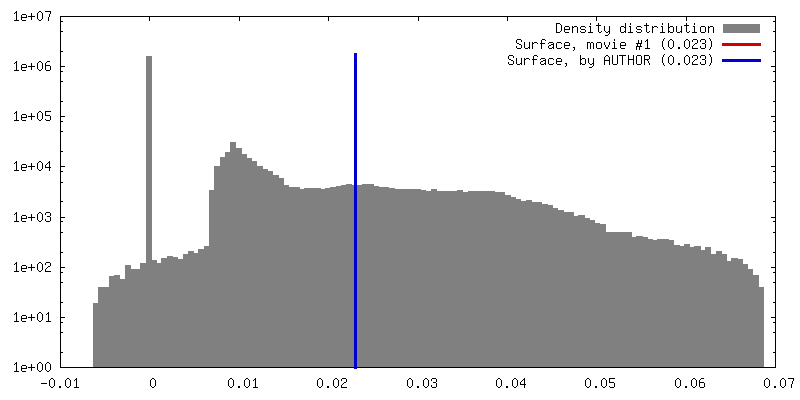
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
- Sample components
Sample components
-Entire : Mature Aminopeptidase-1 dodecamer
| Entire | Name: Mature Aminopeptidase-1 dodecamer |
|---|---|
| Components |
|
-Supramolecule #1: Mature Aminopeptidase-1 dodecamer
| Supramolecule | Name: Mature Aminopeptidase-1 dodecamer / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 600 KDa |
-Macromolecule #1: Vacuolar aminopeptidase 1
| Macromolecule | Name: Vacuolar aminopeptidase 1 / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO / EC number: aminopeptidase I |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 57.16243 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: MEEQREILEQ LKKTLQMLTV EPSKNNQIAN EEKEKKENEN SWCILEHNYE DIAQEFIDFI YKNPTTYHVV SFFAELLDKH NFKYLSEKS NWQDSIGEDG GKFYTIRNGT NLSAFILGKN WRAEKGVGVI GSHVDALTVK LKPVSFKDTA EGYGRIAVAP Y GGTLNELW ...String: MEEQREILEQ LKKTLQMLTV EPSKNNQIAN EEKEKKENEN SWCILEHNYE DIAQEFIDFI YKNPTTYHVV SFFAELLDKH NFKYLSEKS NWQDSIGEDG GKFYTIRNGT NLSAFILGKN WRAEKGVGVI GSHVDALTVK LKPVSFKDTA EGYGRIAVAP Y GGTLNELW LDRDLGIGGR LLYKKKGTNE IKSALVDSTP LPVCRIPSLA PHFGKPAEGP FDKEDQTIPV IGFPTPDEEG NE PPTDDEK KSPLFGKHCI HLLRYVAKLA GVEVSELIQM DLDLFDVQKG TIGGIGKHFL FAPRLDDRLC SFAAMIALIC YAK DVNTEE SDLFSTVTLY DNEEIGSLTR QGAKGGLLES VVERSSSAFT KKPVDLHTVW ANSIILSADV NHLYNPNFPE VYLK NHFPV PNVGITLSLD PNGHMATDVV GTALVEELAR RNGDKVQYFQ IKNNSRSGGT IGPSLASQTG ARTIDLGIAQ LSMHS IRAA TGSKDVGLGV KFFNGFFKHW RSVYDEFGEL UniProtKB: Vacuolar aminopeptidase 1 |
-Experimental details
-Structure determination
| Method | negative staining |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.5 Component:
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Staining | Type: NEGATIVE / Material: Uranyl acetate | |||||||||
| Details | The sample was purified using a GraFix gradient. |
- Electron microscopy
Electron microscopy
| Microscope | FEI/PHILIPS CM12 |
|---|---|
| Image recording | Film or detector model: TVIPS TEMCAM-F415 (4k x 4k) / Average electron dose: 40.0 e/Å2 |
| Electron beam | Acceleration voltage: 120 kV / Electron source: LAB6 |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal magnification: 53000 |
| Sample stage | Specimen holder model: SIDE ENTRY, EUCENTRIC |

