 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-20110 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Separating distinct macromolecular assemblies from cryo-EM images | |||||||||
 マップデータ マップデータ | Reconstruction of the 60S ribosome | |||||||||
 試料 試料 |
| |||||||||
| 生物種 |  | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 4.0 Å | |||||||||
 データ登録者 データ登録者 | Verbeke EJ / Zhou Y / Horton AP / Mallam AL / Taylor DW / Marcotte EM | |||||||||
 引用 引用 | ジャーナル: J Struct Biol / 年: 2020 タイトル: Separating distinct structures of multiple macromolecular assemblies from cryo-EM projections. 著者: Eric J Verbeke / Yi Zhou / Andrew P Horton / Anna L Mallam / David W Taylor / Edward M Marcotte /  要旨: Single particle analysis for structure determination in cryo-electron microscopy is traditionally applied to samples purified to near homogeneity as current reconstruction algorithms are not designed ...Single particle analysis for structure determination in cryo-electron microscopy is traditionally applied to samples purified to near homogeneity as current reconstruction algorithms are not designed to handle heterogeneous mixtures of structures from many distinct macromolecular complexes. We extend on long established methods and demonstrate that relating two-dimensional projection images by their common lines in a graphical framework is sufficient for partitioning distinct protein and multiprotein complexes within the same data set. The feasibility of this approach is first demonstrated on a large set of synthetic reprojections from 35 unique macromolecular structures spanning a mass range of hundreds to thousands of kilodaltons. We then apply our algorithm on cryo-EM data collected from a mixture of five protein complexes and use existing methods to solve multiple three-dimensional structures ab initio. Incorporating methods to sort single particle cryo-EM data from extremely heterogeneous mixtures will alleviate the need for stringent purification and pave the way toward investigation of samples containing many unique structures. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
 ムービービューア ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_20110.map.gz | 108.8 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-20110-v30.xmlemd-20110.xml | 10.7 KB 10.7 KB | 表示 表示 | EMDBヘッダ |
| 画像 | 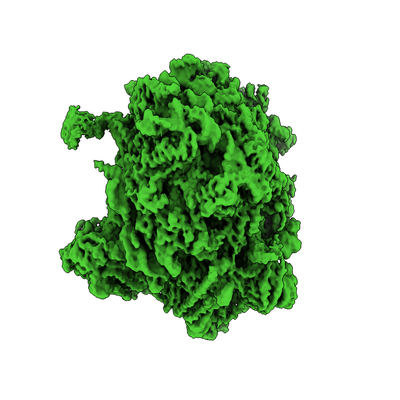 emd_20110.png emd_20110.png | 105.8 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-20110ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20110 http://ftp.pdbj.org/pub/emdb/structures/EMD-20110ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20110 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_20110_validation.pdf.gz | 78.6 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_20110_full_validation.pdf.gz | 77.8 KB | 表示 | |
| XML形式データ | emd_20110_validation.xml.gz | 493 B | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-20110ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-20110 | HTTPS FTP |
-関連構造データ
| 関連構造データ | C: 同じ文献を引用 ( |
|---|---|
| 類似構造データ | |
| 電子顕微鏡画像生データ | EMPIAR-10268 (タイトル: Separating distinct macromolecular assemblies from cryo-EM images Data size: 128.5 Data #1: Drift-corrected and dose-weighted average micographs of a mixture containing 40S, 60S, 80S and apoferritin [micrographs - single frame]) |













-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |

-マップ
| ファイル | ダウンロード / ファイル: emd_20110.map.gz / 形式: CCP4 / 大きさ: 216 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Reconstruction of the 60S ribosome | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-添付データ
- 試料の構成要素
試料の構成要素
-全体 : 60S ribosome
| 全体 | 名称: 60S ribosome |
|---|---|
| 要素 |
|
-超分子 #1: 60S ribosome
| 超分子 | 名称: 60S ribosome / タイプ: complex / ID: 1 / 親要素: 0 / 詳細: From a complex mixture of defined assemblies |
|---|---|
| 由来(天然) | 生物種: |
| 分子量 | 理論値: 2 MDa |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 1 mg/mL |
|---|---|
| 緩衝液 | pH: 7.4 |
| グリッド | 詳細: unspecified |
| 凍結 | 凍結剤: ETHANE / チャンバー内湿度: 100 % / 装置: FEI VITROBOT MARK IV |
| 詳細 | a mixture of 40S, 60S, 80S, and apoferritin |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 撮影 | フィルム・検出器のモデル: GATAN K2 SUMMIT (4k x 4k) 検出モード: COUNTING / デジタル化 - 画像ごとのフレーム数: 1-20 / 撮影したグリッド数: 1 / 実像数: 2400 / 平均露光時間: 6.0 sec. / 平均電子線量: 40.0 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2.7 mm / 最大 デフォーカス(公称値): -3.0 µm / 最小 デフォーカス(公称値): -2.0 µm |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
