 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-13355: Structure of the Caulobacter crescentus S-layer protein RsaA N-te... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-13355 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the Caulobacter crescentus S-layer protein RsaA N-terminal domain bound to LPS and soaked with Holmium | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | S-layer protein RsaA bound to LPS and Holmium / STRUCTURAL PROTEIN | |||||||||
| Function / homology | RsaA N-terminal domain / S-layer / RTX calcium-binding nonapeptide repeat / RTX calcium-binding nonapeptide repeat (4 copies) / Serralysin-like metalloprotease, C-terminal / calcium ion binding / S-layer protein Function and homology information Function and homology information | |||||||||
| Biological species |  Caulobacter vibrioides (bacteria) Caulobacter vibrioides (bacteria) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 4.37 Å | |||||||||
 Authors Authors | von Kugelgen A / Bharat TAM | |||||||||
| Funding support |  United Kingdom, 2 items United Kingdom, 2 items
| |||||||||
 Citation Citation | Journal: Structure / Year: 2022 Title: High-resolution mapping of metal ions reveals principles of surface layer assembly in Caulobacter crescentus cells. Authors: Matthew Herdman / Andriko von Kügelgen / Danguole Kureisaite-Ciziene / Ramona Duman / Kamel El Omari / Elspeth F Garman / Andreas Kjaer / Dimitrios Kolokouris / Jan Löwe / Armin Wagner / ...Authors: Matthew Herdman / Andriko von Kügelgen / Danguole Kureisaite-Ciziene / Ramona Duman / Kamel El Omari / Elspeth F Garman / Andreas Kjaer / Dimitrios Kolokouris / Jan Löwe / Armin Wagner / Phillip J Stansfeld / Tanmay A M Bharat / Abstract: Surface layers (S-layers) are proteinaceous crystalline coats that constitute the outermost component of most prokaryotic cell envelopes. In this study, we have investigated the role of metal ions in ...Surface layers (S-layers) are proteinaceous crystalline coats that constitute the outermost component of most prokaryotic cell envelopes. In this study, we have investigated the role of metal ions in the formation of the Caulobacter crescentus S-layer using high-resolution structural and cell biology techniques, as well as molecular simulations. Utilizing optical microscopy of fluorescently tagged S-layers, we show that calcium ions facilitate S-layer lattice formation and cell-surface binding. We report all-atom molecular dynamics simulations of the S-layer lattice, revealing the importance of bound metal ions. Finally, using electron cryomicroscopy and long-wavelength X-ray diffraction experiments, we mapped the positions of metal ions in the S-layer at near-atomic resolution, supporting our insights from the cellular and simulations data. Our findings contribute to the understanding of how C. crescentus cells form a regularly arranged S-layer on their surface, with implications on fundamental S-layer biology and the synthetic biology of self-assembling biomaterials. | |||||||||
| History |
|
- Structure visualization
Structure visualization
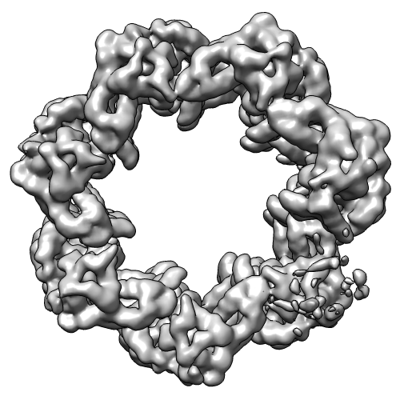
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_13355.map.gz | 80.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-13355-v30.xmlemd-13355.xml | 24.1 KB 24.1 KB | Display Display | EMDB header |
| Images |  emd_13355.png emd_13355.png | 114.7 KB | ||
| Filedesc metadata | emd-13355.cif.gz | 8.4 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-13355ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13355 http://ftp.pdbj.org/pub/emdb/structures/EMD-13355ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13355 | HTTPS FTP |
-Related structure data
| Related structure data |  7peoMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10790 (Title: High-resolution mapping of metal ions reveals principles of surface layer assembly in Caulobacter crescentus cells Data size: 304.4 Data #1: Unaligned multiframe micrographs of RsaA_NTD spiral soaked with 5 mM HoCl3 for 2 hours (converted from .mrc into .tif with relion_convert_to_tiff) [micrographs - multiframe] Data #2: Unaligned multiframe micrographs of RsaA_NTD spiral soaked with 5 mM HoCl3 for 2 hours with a 30 degree stage tilt (converted from .mrc into .tif with relion_convert_to_tiff) [micrographs - multiframe] Data #3: Aligned and dose-weighted micrographs of RsaA_NTD spiral soaked with 5 mM HoCl3 for 2 hours [micrographs - single frame] Data #4: Aligned and dose-weighted micrographs of RsaA_NTD spiral soaked with 5 mM HoCl3 for 2 hours with a 30 degree stage tilt [micrographs - single frame] Data #5: Gain reference image of the non-tilted and tilted dataset in MRC format [micrographs - single frame]) |





-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_13355.map.gz / Format: CCP4 / Size: 103 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.08 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
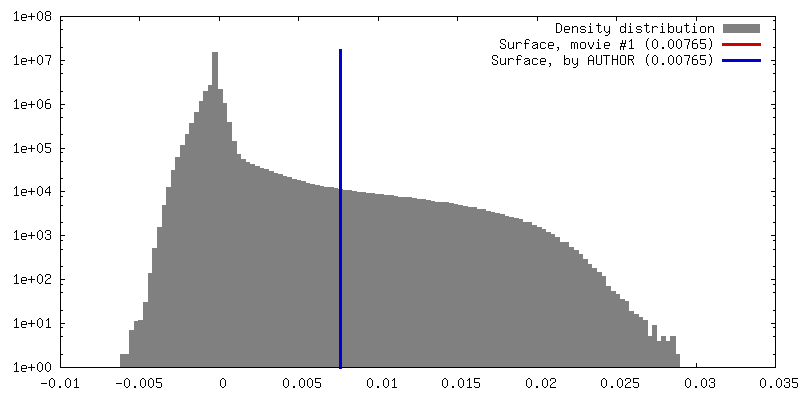
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
- Sample components
Sample components
-Entire : Structure of the Caulobacter crescentus S-layer protein RsaA N-te...
| Entire | Name: Structure of the Caulobacter crescentus S-layer protein RsaA N-terminal domain bound to LPS and soaked with Holmium |
|---|---|
| Components |
|
-Supramolecule #1: Structure of the Caulobacter crescentus S-layer protein RsaA N-te...
| Supramolecule | Name: Structure of the Caulobacter crescentus S-layer protein RsaA N-terminal domain bound to LPS and soaked with Holmium type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 Details: Structure of the Caulobacter crescentus S-layer protein RsaA N-terminal domain bound to LPS and soaked with Holmium |
|---|---|
| Source (natural) | Organism: Caulobacter vibrioides (bacteria) / Strain: YB1001 |
-Macromolecule #1: S-layer protein
| Macromolecule | Name: S-layer protein / type: protein_or_peptide / ID: 1 / Details: LPS O-antigen bound to the protein / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Caulobacter vibrioides (bacteria) / Strain: YB1001 |
| Molecular weight | Theoretical: 25.820354 KDa |
| Recombinant expression | Organism: Caulobacter vibrioides CB15 (bacteria) |
| Sequence | String: AYTTAQLVTA YTNANLGKAP DAATTLTLDA YATQTQTGGL SDAAALTNTL KLVNSTTAVA IQTYQFFTGV APSAAGLDFL VDSTTNTND LNDAYYSKFA QENRFINFSI NLATGAGAGA TAFAAAYTGV SYAQTVATAY DKIIGNAVAT AAGVDVAAAV A FLSRQANI ...String: AYTTAQLVTA YTNANLGKAP DAATTLTLDA YATQTQTGGL SDAAALTNTL KLVNSTTAVA IQTYQFFTGV APSAAGLDFL VDSTTNTND LNDAYYSKFA QENRFINFSI NLATGAGAGA TAFAAAYTGV SYAQTVATAY DKIIGNAVAT AAGVDVAAAV A FLSRQANI DYLTAFVRAN TPFTAAADID LAVKAALIGT ILNAATVSGI GGYATATAAM INDLSDGALS TDNAAGVNLF TA YPSSGVS GSENLYFQ UniProtKB: S-layer protein |
-Macromolecule #3: CALCIUM ION
| Macromolecule | Name: CALCIUM ION / type: ligand / ID: 3 / Number of copies: 2 / Formula: CA |
|---|---|
| Molecular weight | Theoretical: 40.078 Da |
-Macromolecule #4: HOLMIUM ATOM
| Macromolecule | Name: HOLMIUM ATOM / type: ligand / ID: 4 / Number of copies: 1 / Formula: HO |
|---|---|
| Molecular weight | Theoretical: 164.93 Da |
| Chemical component information |  ChemComp-HO: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 2.25 mg/mL | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
Details: Buffer solutions were prepared fresh from sterile filtered concentrated stocksolutions. Solutions were filtered through a 0.22 um filter to avoid microbial contamination and degassed using a ...Details: Buffer solutions were prepared fresh from sterile filtered concentrated stocksolutions. Solutions were filtered through a 0.22 um filter to avoid microbial contamination and degassed using a vacuum fold pump. The pH of the HEPES stock solution was adjusted with sodium hydroxide at 4 deg C. 5 mM HoCl3 was added to the specimen 1.5 hours before vitrification. | ||||||||||||||||||
| Grid | Model: Quantifoil R2/2 / Material: COPPER/RHODIUM / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 20 sec. / Pretreatment - Atmosphere: AIR / Details: 20 seconds, 15 mA | ||||||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 283.15 K / Instrument: FEI VITROBOT MARK IV Details: Vitrobot options: Blot time 4 seconds, Blot force -13,1, Wait time 10 seconds, Drain time 0.5 seconds,. | ||||||||||||||||||
| Details | RsaA N-terminal domain with LPS soaked with 5 mM HoCl3 for 1.5 h on ice before vitrification |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 70.0 K / Max: 70.0 K |
| Specialist optics | Energy filter - Name: GIF Quantum LS / Energy filter - Slit width: 20 eV |
| Details | EPU software |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3838 pixel / Digitization - Dimensions - Height: 3710 pixel / Digitization - Frames/image: 1-20 / Number grids imaged: 2 / Number real images: 2038 / Average exposure time: 8.0 sec. / Average electron dose: 44.8 e/Å2 Details: Two data collections: First: 0 degree stage tilt with 903 collected movies. Second: 30 degree stage tilt with 1135 collected movies |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Calibrated defocus max: -4.0 µm / Calibrated defocus min: -1.0 µm / Calibrated magnification: 130000 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: -4.0 µm / Nominal defocus min: -1.0 µm / Nominal magnification: 130000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Chain ID: A / Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Details | The atomic coordinates (PDB ID 6T72) of our previous cryo-EM structure (von Kugelgen et al., 2020) of the RsaANTD oligomer bound to the O-antigen of lipopolysaccharide (LPS) were rigid body fitted into the final post-processed map from Relion 3.0 (Zivanov et al., 2018) using UCSF Chimera (Pettersen et al., 2004). The resulting fitted model was subjected to real-space refinement using Refmac5 (Murshudov et al., 2011) inside the CCP-EM suite (Burnely et al., 2017), as described previously (von Kugelgen et al., 2020), using reference restraints of the initial structure (PDB ID 6T72) generated with PROSMART (Nicholls et al. 2012). |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
| Output model | PDB-7peo: |

