 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4ky8: Crystal structure of TS-DHFR from Cryptosporidium hominis in comp... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4ky8 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of TS-DHFR from Cryptosporidium hominis in complex with NADPH, methotrexate, FdUMP and 4-((2-amino-6-methyl-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)thio)-2-chlorophenyl)-L-glutamic acid | ||||||
 Components Components | Bifunctional thymidylate synthase-dihydrofolate reductase | ||||||
 Keywords Keywords | TRANSFERASE / OXIDOREDUCTASE/INHIBITOR / bifunctional enzyme / oxidoreductase / OXIDOREDUCTASE-INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationThymidylate Synthase; Chain A / Thymidylate synthase/dCMP hydroxymethylase domain / Dihydrofolate Reductase, subunit A / Dihydrofolate Reductase, subunit A / 2-Layer Sandwich / 3-Layer(aba) Sandwich / Alpha Beta Similarity search - Domain/homology | ||||||
| Biological species |  Cryptosporidium hominis (eukaryote) Cryptosporidium hominis (eukaryote) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.084 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.084 Å | ||||||
 Authors Authors | Kumar, V.P. / Anderson, K.S. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2013 Title: Substituted pyrrolo[2,3-d]pyrimidines as Cryptosporidium hominis thymidylate synthase inhibitors. Authors: Kumar, V.P. / Frey, K.M. / Wang, Y. / Jain, H.K. / Gangjee, A. / Anderson, K.S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4ky8.cif.gz | 524.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4ky8.ent.gz | 435 KB | Display | PDB format |
| PDBx/mmJSON format | 4ky8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ky/4ky8ftp://data.pdbj.org/pub/pdb/validation_reports/ky/4ky8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1qzfS S: Starting model for refinement |
|---|---|
| Similar structure data |
























-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 60262.520 Da / Num. of mol.: 5 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Cryptosporidium hominis (eukaryote) / Gene: Chro.40506, TS-DHFR / Plasmid: pTrc99A / Production host:  References: UniProt: Q5CGA3, thymidylate synthase, dihydrofolate reductase #2: Chemical | ChemComp-NDP / 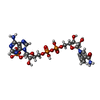  Mass: 745.421 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C21H30N7O17P3 Mass: 745.421 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C21H30N7O17P3#3: Chemical | ChemComp-UFP / 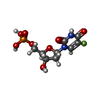  Type: DNA linking / Mass: 326.172 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C9H12FN2O8P Type: DNA linking / Mass: 326.172 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C9H12FN2O8P#4: Chemical | ChemComp-MTX / 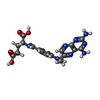  Mass: 454.439 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C20H22N8O5 Mass: 454.439 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C20H22N8O5#5: Chemical | ChemComp-1UF /   Mass: 479.894 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C19H18ClN5O6S Mass: 479.894 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C19H18ClN5O6S |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.48 Å3/Da / Density % sol: 72.56 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 8 Details: 14% (w/v) PEG 6000, 60 mM ammonium sulfate, 200 mM lithium sulfate and 100 mM Tris pH 8.0. ChTS-DHFR enzyme (~ 7 mg/ml) with 1 mM NADPH, 1 mM MTX, 1 mM FdUMP and 0.5 mM inhibitor., VAPOR ...Details: 14% (w/v) PEG 6000, 60 mM ammonium sulfate, 200 mM lithium sulfate and 100 mM Tris pH 8.0. ChTS-DHFR enzyme (~ 7 mg/ml) with 1 mM NADPH, 1 mM MTX, 1 mM FdUMP and 0.5 mM inhibitor., VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 77 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X25 / Wavelength: 1.1 Å / Beamline: X25 / Wavelength: 1.1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Apr 16, 2013 / Details: monochromator | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Double silicon (111) crystal monochromator with cryogenically-cooled first crystal and sagittally-bent second crystal horizontally focusing at 3.3:1 demagnification Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 3.08→48.12 Å / Num. obs: 70173 / % possible obs: 71.6 % / Observed criterion σ(F): 1.5 / Redundancy: 3 % / Biso Wilson estimate: 70.12 Å2 / Rmerge(I) obs: 0.207 / Rsym value: 0.207 / Net I/σ(I): 4.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1QZF Resolution: 3.084→48.12 Å / Occupancy max: 1 / Occupancy min: 1 / SU ML: 0.39 / σ(F): 1.34 / Phase error: 29.7 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 82.3969 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.084→48.12 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
