 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3smi: Human poly(ADP-ribose) polymerase 14 (Parp14/Artd8) - catalytic d... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3smi | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human poly(ADP-ribose) polymerase 14 (Parp14/Artd8) - catalytic domain in complex with a quinazoline inhibitor | ||||||
 Components Components | Poly [ADP-ribose] polymerase 14 | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE Inhibitor / DIPHTHERIA TOXIN LIKE FOLD / TRANSFERASE / NAD+ / ADP-RIBOSYLATION / TRANSFERASE-TRANSFERASE Inhibitor complex / Structural Genomics / Structural Genomics Consortium / SGC | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of interleukin-4-mediated signaling pathway / negative regulation of tyrosine phosphorylation of STAT protein / Nicotinamide salvaging / Maturation of nucleoprotein / Maturation of nucleoprotein / protein poly-ADP-ribosylation / negative regulation of type II interferon-mediated signaling pathway / NAD+-protein ADP-ribosyltransferase activity / Transferases; Glycosyltransferases; Pentosyltransferases / NAD+-protein poly-ADP-ribosyltransferase activity ...positive regulation of interleukin-4-mediated signaling pathway / negative regulation of tyrosine phosphorylation of STAT protein / Nicotinamide salvaging / Maturation of nucleoprotein / Maturation of nucleoprotein / protein poly-ADP-ribosylation / negative regulation of type II interferon-mediated signaling pathway / NAD+-protein ADP-ribosyltransferase activity / Transferases; Glycosyltransferases; Pentosyltransferases / NAD+-protein poly-ADP-ribosyltransferase activity / NAD+ binding / positive regulation of tyrosine phosphorylation of STAT protein / nucleotidyltransferase activity / transcription corepressor activity / negative regulation of gene expression / innate immune response / enzyme binding / membrane / nucleus / plasma membrane / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Karlberg, T. / Moche, M. / Arrowsmith, C.H. / Berglund, H. / Bountra, C. / Edwards, A.M. / Ekblad, T. / Graslund, S. / Kouznetsova, E. / Nordlund, P. ...Karlberg, T. / Moche, M. / Arrowsmith, C.H. / Berglund, H. / Bountra, C. / Edwards, A.M. / Ekblad, T. / Graslund, S. / Kouznetsova, E. / Nordlund, P. / Nyman, T. / Thorsell, A.G. / Tresaugues, L. / Weigelt, J. / Schuler, H. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Nat.Biotechnol. / Year: 2012 Title: Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Authors: Wahlberg, E. / Karlberg, T. / Kouznetsova, E. / Markova, N. / Macchiarulo, A. / Thorsell, A.G. / Pol, E. / Frostell, A. / Ekblad, T. / Oncu, D. / Kull, B. / Robertson, G.M. / Pellicciari, R. ...Authors: Wahlberg, E. / Karlberg, T. / Kouznetsova, E. / Markova, N. / Macchiarulo, A. / Thorsell, A.G. / Pol, E. / Frostell, A. / Ekblad, T. / Oncu, D. / Kull, B. / Robertson, G.M. / Pellicciari, R. / Schuler, H. / Weigelt, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3smi.cif.gz | 87.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3smi.ent.gz | 66.3 KB | Display | PDB format |
| PDBx/mmJSON format | 3smi.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 3smi_validation.pdf.gz | 929.1 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 3smi_full_validation.pdf.gz | 933.4 KB | Display | |
| Data in XML | 3smi_validation.xml.gz | 15.7 KB | Display | |
| Data in CIF | 3smi_validation.cif.gz | 20.9 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sm/3smiftp://data.pdbj.org/pub/pdb/validation_reports/sm/3smi | HTTPS FTP |
-Related structure data
| Related structure data |  3goySC  3mhjC  3mhkC  3p0nC  3p0pC  3p0qC  3se2C 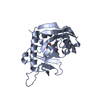 3smjC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22118.590 Da / Num. of mol.: 2 / Fragment: Catalytic domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: BAL2, KIAA1268, PARP14 / Plasmid: pNIC-Bsa4 / Production host:  #2: Chemical | 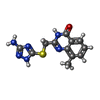  Mass: 288.328 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H12N6OS Mass: 288.328 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H12N6OS#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 61 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 61 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.21 Å3/Da / Density % sol: 44.34 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 25% PEG3350, 0.2M NaNO3, pH 7.5, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.91841 Å / Beamline: 14.1 / Wavelength: 0.91841 Å |
| Detector | Type: RAYONIX MX-225 / Detector: CCD / Date: Aug 21, 2010 / Details: mirrors and double crystal monochromator |
| Radiation | Monochromator: double crystals Si-111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.91841 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→35 Å / Num. all: 16003 / Num. obs: 16003 / % possible obs: 99.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.4 % / Biso Wilson estimate: 39.21 Å2 / Rmerge(I) obs: 0.109 / Rsym value: 0.179 / Net I/σ(I): 12.6 |
| Reflection shell | Resolution: 2.4→2.46 Å / Redundancy: 4.5 % / Rmerge(I) obs: 0.441 / Mean I/σ(I) obs: 3.8 / Num. unique all: 1155 / Rsym value: 0.462 / % possible all: 99.9 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3GOY Resolution: 2.4→33.67 Å / Cor.coef. Fo:Fc: 0.9057 / Cor.coef. Fo:Fc free: 0.872 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 42.21 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.362 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→33.67 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.57 Å / Total num. of bins used: 8
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
