CD209 (DC-SIGN) signaling / HDL assembly / Regulation of insulin secretion / Rap1 signalling / Ion homeostasis / PKA activation in glucagon signalling / DARPP-32 events / CREB1 phosphorylation through the activation of Adenylate Cyclase / GPER1 signaling / Factors involved in megakaryocyte development and platelet production ...CD209 (DC-SIGN) signaling / HDL assembly / Regulation of insulin secretion / Rap1 signalling / Ion homeostasis / PKA activation in glucagon signalling / DARPP-32 events / CREB1 phosphorylation through the activation of Adenylate Cyclase / GPER1 signaling / Factors involved in megakaryocyte development and platelet production / Loss of Nlp from mitotic centrosomes / Recruitment of mitotic centrosome proteins and complexes / Loss of proteins required for interphase microtubule organization from the centrosome / Recruitment of NuMA to mitotic centrosomes / Anchoring of the basal body to the plasma membrane / AURKA Activation by TPX2 / RET signaling / Interleukin-3, Interleukin-5 and GM-CSF signaling / VEGFA-VEGFR2 Pathway / Regulation of PLK1 Activity at G2/M Transition / PKA activation / Hedgehog 'off' state / MAPK6/MAPK4 signaling / negative regulation of cAMP-dependent protein kinase activity / GLI3 is processed to GLI3R by the proteasome / dorsal/ventral neural tube patterning / regulation of protein processing / cAMP-dependent protein kinase inhibitor activity / cellular response to parathyroid hormone stimulus / protein localization to lipid droplet / histone H1-4S35 kinase activity / cAMP-dependent protein kinase / cAMP-dependent protein kinase activity / regulation of G2/M transition of mitotic cell cycle / mesoderm formation / negative regulation of protein import into nucleus / regulation of bicellular tight junction assembly / cAMP-dependent protein kinase complex / cellular response to cold / interleukin-2-mediated signaling pathway / Mitochondrial protein degradation / sperm capacitation / regulation of osteoblast differentiation / Glucagon-like Peptide-1 (GLP1) regulates insulin secretion / negative regulation of glycolytic process through fructose-6-phosphate / High laminar flow shear stress activates signaling by PIEZO1 and PECAM1:CDH5:KDR in endothelial cells / Vasopressin regulates renal water homeostasis via Aquaporins / protein kinase A regulatory subunit binding / ciliary base / protein kinase A catalytic subunit binding / TORC1 signaling / intracellular potassium ion homeostasis / plasma membrane raft / neural tube closure / axoneme / sperm flagellum / postsynaptic modulation of chemical synaptic transmission / negative regulation of cAMP/PKA signal transduction / negative regulation of TORC1 signaling / protein export from nucleus / positive regulation of gluconeogenesis / protein serine/threonine/tyrosine kinase activity / cellular response to glucagon stimulus / cellular response to nutrient levels / positive regulation of phagocytosis / positive regulation of protein export from nucleus / acrosomal vesicle / regulation of proteasomal protein catabolic process / negative regulation of smoothened signaling pathway / cellular response to glucose stimulus / neuromuscular junction / positive regulation of insulin secretion / positive regulation of cholesterol biosynthetic process / mRNA processing / adenylate cyclase-inhibiting G protein-coupled receptor signaling pathway / manganese ion binding / cellular response to heat / adenylate cyclase-activating G protein-coupled receptor signaling pathway / regulation of cell cycle / protein kinase activity / postsynapse / nuclear speck / protein domain specific binding / protein serine kinase activity / protein serine/threonine kinase activity / centrosome / ubiquitin protein ligase binding / protein kinase binding / perinuclear region of cytoplasm / negative regulation of transcription by RNA polymerase II / glutamatergic synapse / magnesium ion binding / mitochondrion / ATP binding / nucleus / cytosol / cytoplasm Similarity search - Function
cAMP-dependent protein kinase inhibitor / cAMP-dependent protein kinase inhibitor / cAMP-dependent protein kinase catalytic subunit / Extension to Ser/Thr-type protein kinases / AGC-kinase, C-terminal / AGC-kinase C-terminal domain profile. / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 ...cAMP-dependent protein kinase inhibitor / cAMP-dependent protein kinase inhibitor / cAMP-dependent protein kinase catalytic subunit / Extension to Ser/Thr-type protein kinases / AGC-kinase, C-terminal / AGC-kinase C-terminal domain profile. / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / 2-Layer Sandwich / Orthogonal Bundle / Mainly Alpha / Alpha Beta Similarity search - Domain/homology
1-[1-(9H-purin-6-yl)piperidin-4-yl]methanamine / cAMP-dependent protein kinase catalytic subunit alpha / cAMP-dependent protein kinase inhibitor alpha Similarity search - Component
Biological species
BOS TAURUS (domestic cattle) HOMO SAPIENS (human)
Method
X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.09 Å
Mass: 18.015 Da / Num. of mol.: 391 / Source method: isolated from a natural source / Formula: H2O
Compound details
ENGINEERED RESIDUE IN CHAIN A, VAL 105 TO THR ENGINEERED RESIDUE IN CHAIN A, VAL 124 TO ALA ...ENGINEERED RESIDUE IN CHAIN A, VAL 105 TO THR ENGINEERED RESIDUE IN CHAIN A, VAL 124 TO ALA ENGINEERED RESIDUE IN CHAIN A, LEU 174 TO MET ENGINEERED RESIDUE IN CHAIN A, GLN 182 TO LYS
Has protein modification
Y
Sequence details
4 RESIDUES OF THE WILD-TYPE PKA SEQUENCE HAVE BEEN MUTATED TO THE CORRESPONDING RESIDUES IN PKB
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.31 Å3/Da / Density % sol: 46.26 % / Description: NONE
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads














































































 PDBj
PDBj



 Assembly
Assembly

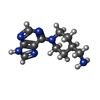
 Mass: 232.285 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H16N6
Mass: 232.285 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H16N6 Mass: 18.015 Da / Num. of mol.: 391 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 391 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing