 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1qj3: Crystal structure of 7,8-diaminopelargonic acid synthase in compl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qj3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of 7,8-diaminopelargonic acid synthase in complex with 7-keto-8-aminopelargonic acid | ||||||
 Components Components | 7,8-DIAMINOPELARGONIC ACID SYNTHASE | ||||||
 Keywords Keywords | TRANSFERASE / AMINOTRANSFERASE / PYRIDOXAL-5'-PHOSPHATE / BIOTIN BIOSYNTHESIS | ||||||
| Function / homology |  Function and homology information Function and homology informationadenosylmethionine-8-amino-7-oxononanoate transaminase / S-adenosyl-L-methionine:8-amino-7-oxononanoate transaminase activity / biotin biosynthetic process / pyridoxal phosphate binding / protein homodimerization activity / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / OTHER / Resolution: 2.7 Å X-RAY DIFFRACTION / OTHER / Resolution: 2.7 Å | ||||||
 Authors Authors | Kaeck, H. / Sandmark, J. / Gibson, K.J. / Lindqvist, Y. / Schneider, G. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1999 Title: Crystal Structure of Diaminopelargonic Acid Synthase; Evolutionary Relationships between Pyridoxal-5'-Phosphate Dependent Enzymes Authors: Kack, H. / Sandmark, J. / Gibson, K.J. / Schneider, G. / Lindqvist, Y. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qj3.cif.gz | 172.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qj3.ent.gz | 136.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1qj3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qj/1qj3ftp://data.pdbj.org/pub/pdb/validation_reports/qj/1qj3 | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.99995, 0.01019, 0.0007), Vector: |
-Components
| #1: Protein | Mass: 47310.391 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P12995, adenosylmethionine-8-amino-7-oxononanoate transaminase #2: Chemical |   Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P#3: Chemical | 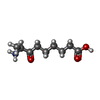  Mass: 187.236 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H17NO3 Mass: 187.236 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H17NO3#4: Chemical |   Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 94 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 94 / Source method: isolated from a natural source / Formula: H2OHas protein modification | N | Nonpolymer details | PYRIDOXAL-5'-PHOSPHATE IS BOUND TO LYS 274 IN BOTH CHAINS | Sequence details | FROM THE ELECTRON DENSITY TRP14 IS REINTERPRE | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.06 Å3/Da / Density % sol: 30 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / pH: 7.5 Details: 25% PEG-4000, 20% MPD, 100MM HEPES PH7.5, 5MM KAPA (7-KETO-8-AMINOPELARGONIC ACID), 20 DEGREES C, MICROSEEDING., pH 7.50 | ||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7.3 / Method: vapor diffusionDetails: drop consists of equal volume of protein and precipitant solutions | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-D / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Oct 15, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→20 Å / Num. obs: 21479 / % possible obs: 99.9 % / Observed criterion σ(I): 2 / Redundancy: 5.8 % / Biso Wilson estimate: 42.6 Å2 / Rsym value: 0.104 / Net I/σ(I): 17.6 |
| Reflection shell | Resolution: 2.7→2.82 Å / Mean I/σ(I) obs: 5.4 / Rsym value: 0.326 / % possible all: 99.7 |
| Reflection | *PLUS Num. measured all: 124908 / Rmerge(I) obs: 0.104 |
| Reflection shell | *PLUS % possible obs: 99.7 % / Rmerge(I) obs: 0.326 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER / Resolution: 2.7→20 Å / Cross valid method: THROUGHOUT / σ(F): 0 Details: DUE TO DISORDERED MAINCHAIN THE FOLLOWING RESIDUES WERE REMOVED FROM THE MODEL OF CHAIN A: SER 160, MET 161, HIS 162, SER 163, LEU 164, TRP 165, LYS 166, GLY 167, TYR 168, ASP 183, GLY 300, ...Details: DUE TO DISORDERED MAINCHAIN THE FOLLOWING RESIDUES WERE REMOVED FROM THE MODEL OF CHAIN A: SER 160, MET 161, HIS 162, SER 163, LEU 164, TRP 165, LYS 166, GLY 167, TYR 168, ASP 183, GLY 300, ALA 301, GLN 429 DUE TO DISORDERED MAINCHAIN THE FOLLOWING RESIDUES WERE REMOVED FROM THE MODEL OF CHAIN B: SER 160, MET 161, HIS 162, SER 163, LEU 164, TRP 165, LYS 166, GLY 167, TYR 168, ASP 183, GLY 184, GLY 300, ALA 301, GLN 429
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 51.8 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
