 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-1080 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | A 11.5 A single particle reconstruction of GroEL using EMAN. | |||||||||
 Map data Map data | This is the volumetric data for our published 11.5 A resolution reconstruction of GroEL. Note that newer EMAN releases can refine the same data to ~9 A resolution. | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 11.5 Å | |||||||||
 Authors Authors | Ludtke SJ / Jakana J / Song J / Chuang DT / Chiu W | |||||||||
 Citation Citation | Journal: J Mol Biol / Year: 2001 Title: A 11.5 A single particle reconstruction of GroEL using EMAN. Authors: S J Ludtke / J Jakana / J L Song / D T Chuang / W Chiu /  Abstract: Single-particle analysis has become an increasingly important method for structural determination of large macromolecular assemblies. GroEL is an 800 kDa molecular chaperone, which, along with its co- ...Single-particle analysis has become an increasingly important method for structural determination of large macromolecular assemblies. GroEL is an 800 kDa molecular chaperone, which, along with its co-chaperonin GroES, promotes protein folding both in vitro and in the bacterial cell. EMAN is a single-particle analysis software package, which was first publicly distributed in 2000. We present a three-dimensional reconstruction of native naked GroEL to approximately 11.5 A performed entirely with EMAN. We demonstrate that the single-particle reconstruction, X-ray scattering data and X-ray crystal structure all agree well at this resolution. These results validate the specific methods of image restoration, reconstruction and evaluation techniques implemented in EMAN. It also demonstrates that the single-particle reconstruction technique and X-ray crystallography will yield consistent structure factors, even at low resolution, when image restoration is performed correctly. A detailed comparison of the single-particle and X-ray structures exhibits some small variations in the equatorial domain of the molecule, likely due to the absence of crystal packing forces in the single-particle reconstruction. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
 UCSF Chimera
UCSF Chimera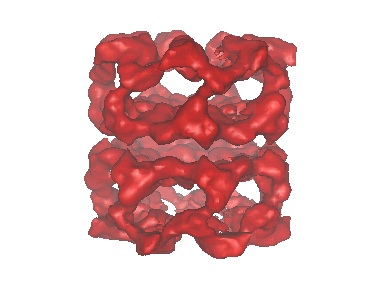
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_1080.map.gz | 618.8 KB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-1080-v30.xmlemd-1080.xml | 9 KB 9 KB | Display Display | EMDB header |
| Images |  1080.gif 1080.gif | 30.8 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-1080ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1080 http://ftp.pdbj.org/pub/emdb/structures/EMD-1080ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1080 | HTTPS FTP |
-Validation report
| Summary document | emd_1080_validation.pdf.gz | 220.6 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_1080_full_validation.pdf.gz | 219.8 KB | Display | |
| Data in XML | emd_1080_validation.xml.gz | 5.2 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-1080ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-1080 | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_1080.map.gz / Format: CCP4 / Size: 3.7 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | This is the volumetric data for our published 11.5 A resolution reconstruction of GroEL. Note that newer EMAN releases can refine the same data to ~9 A resolution. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2.7 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
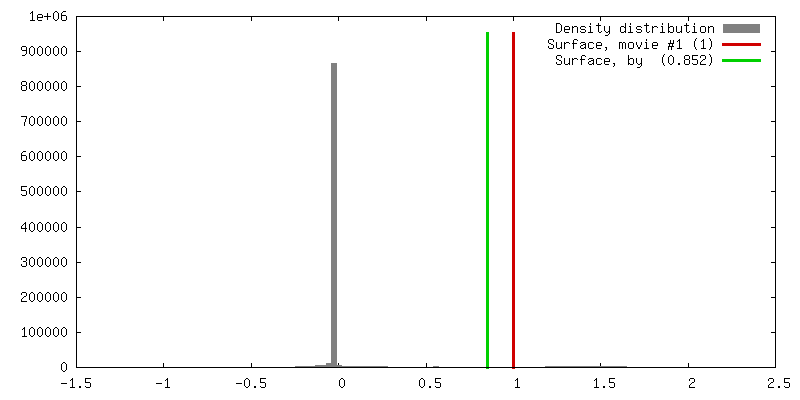
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
- Sample components
Sample components
-Entire : native naked GroEL
| Entire | Name: native naked GroEL |
|---|---|
| Components |
|
-Supramolecule #1000: native naked GroEL
| Supramolecule | Name: native naked GroEL / type: sample / ID: 1000 / Oligomeric state: homo 14-mer / Number unique components: 1 |
|---|---|
| Molecular weight | Theoretical: 800 KDa |
-Macromolecule #1: GroEL
| Macromolecule | Name: GroEL / type: protein_or_peptide / ID: 1 / Number of copies: 14 / Oligomeric state: 14-mer / Recombinant expression: Yes |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Experimental: 57 KDa |
| Recombinant expression | Organism: ESts CG-712 / Recombinant plasmid: pGroESL |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.0 mg/mL |
|---|---|
| Vitrification | Cryogen name: ETHANE / Instrument: HOMEMADE PLUNGER / Details: Vitrification instrument: Manual, gravity, plunger |
- Electron microscopy
Electron microscopy
| Microscope | JEOL 4000EX |
|---|---|
| Image recording | Category: FILM / Film or detector model: KODAK SO-163 FILM / Digitization - Scanner: ZEISS SCAI / Digitization - Sampling interval: 7 µm / Number real images: 5 / Average electron dose: 30 e/Å2 Details: Data was median filtered to 14 microns/pixel after scanning. Bits/pixel: 8 |
| Electron beam | Acceleration voltage: 400 kV / Electron source: LAB6 |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 4.1 mm / Nominal defocus max: 2.4 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 50000 |
| Sample stage | Specimen holder: Side entry / Specimen holder model: GATAN LIQUID NITROGEN |
-Image processing
| Details | Note that class averages are EMAN-style reference-based, not MSA. |
|---|---|
| CTF correction | Details: Per particle phase flipping, amplitude correction during averaging |
| Final reconstruction | Applied symmetry - Point group: D7 (2x7 fold dihedral) / Resolution.type: BY AUTHOR / Resolution: 11.5 Å / Resolution method: FSC 0.5 CUT-OFF / Software - Name: EMAN / Number images used: 4916 |
| Final two d classification | Number classes: 334 |
-Atomic model buiding 1
| Initial model | PDB ID: |
|---|---|
| Software | Name: foldhunter EMAN |
| Details | Protocol: Rigid Body |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: correlation coefficient |

