 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-0175: Helical part of the influenza A virus ribonucleoprotein. Conforma... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-0175 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
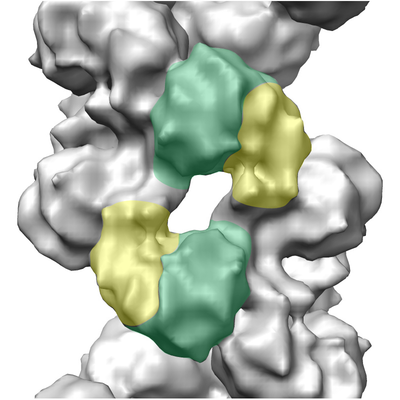
| Title | Helical part of the influenza A virus ribonucleoprotein. Conformation 1. | |||||||||
 Map data Map data | Helical part of influenza A virus ribonucleoprotein (vRNP) | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Influenza A virus Ribonucleoprotein RNA binding protein / VIRAL PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationnegative stranded viral RNA replication / helical viral capsid / viral penetration into host nucleus / host cell / viral nucleocapsid / ribonucleoprotein complex / symbiont entry into host cell / host cell nucleus / structural molecule activity / RNA binding / identical protein binding Similarity search - Function | |||||||||
| Biological species |   Influenza A virus / Influenza A virus (strain A/Wilson-Smith/1933 H1N1) Influenza A virus / Influenza A virus (strain A/Wilson-Smith/1933 H1N1) | |||||||||
| Method | helical reconstruction / cryo EM / Resolution: 11.0 Å | |||||||||
 Authors Authors | Coloma R / Arranz R | |||||||||
| Funding support |  Spain, 2 items Spain, 2 items
| |||||||||
 Citation Citation | Journal: Nat Microbiol / Year: 2020 Title: Structural insights into influenza A virus ribonucleoproteins reveal a processive helical track as transcription mechanism. Authors: Rocío Coloma / Rocío Arranz / José M de la Rosa-Trevín / Carlos O S Sorzano / Sandie Munier / Diego Carlero / Nadia Naffakh / Juan Ortín / Jaime Martín-Benito /   Abstract: The influenza virus genome consists of eight viral ribonucleoproteins (vRNPs), each consisting of a copy of the polymerase, one of the genomic RNA segments and multiple copies of the nucleoprotein ...The influenza virus genome consists of eight viral ribonucleoproteins (vRNPs), each consisting of a copy of the polymerase, one of the genomic RNA segments and multiple copies of the nucleoprotein arranged in a double helical conformation. vRNPs are macromolecular machines responsible for messenger RNA synthesis and genome replication, that is, the formation of progeny vRNPs. Here, we describe the structural basis of the transcription process. The mechanism, which we call the 'processive helical track', is based on the extreme flexibility of the helical part of the vRNP that permits a sliding movement between both antiparallel nucleoprotein-RNA strands, thereby allowing the polymerase to move over the genome while bound to both RNA ends. Accordingly, we demonstrate that blocking this movement leads to inhibition of vRNP transcriptional activity. This mechanism also reveals a critical role of the nucleoprotein in maintaining the double helical structure throughout the copying process to make the RNA template accessible to the polymerase. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_0175.map.gz | 6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-0175-v30.xmlemd-0175.xml | 15.7 KB 15.7 KB | Display Display | EMDB header |
| Images | 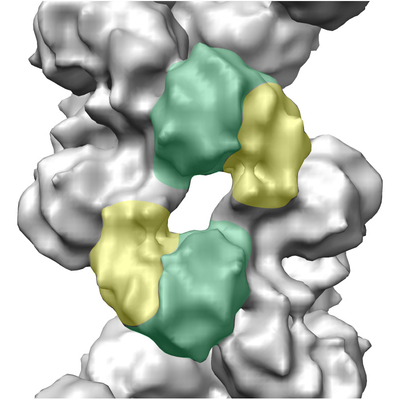 emd_0175.png emd_0175.png | 224.2 KB | ||
| Filedesc metadata | emd-0175.cif.gz | 6.2 KB | ||
| Others | emd_0175_additional.map.gz | 2.8 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-0175ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0175 http://ftp.pdbj.org/pub/emdb/structures/EMD-0175ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0175 | HTTPS FTP |
-Related structure data
| Related structure data |  6h9gMC  4412C  4423C  4426C  4430C  6i54C  6i7bC  6i7mC  6i85C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |



-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_0175.map.gz / Format: CCP4 / Size: 6.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Helical part of influenza A virus ribonucleoprotein (vRNP) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2.26 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Additional map: MonoRes 3D Resolution map. colorkey 0.01,0.05 0.02,0.95 7.00...
| File | emd_0175_additional.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | MonoRes 3D Resolution map. colorkey 0.01,0.05 0.02,0.95 7.00 #000080 " " #0018ff 12.0 #004cff " " #0080ff " " #00e4f8 16.0 #29ffce " " #a4ff53 21.0 #ceff29 " " #bb0000 30.0 #800000 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






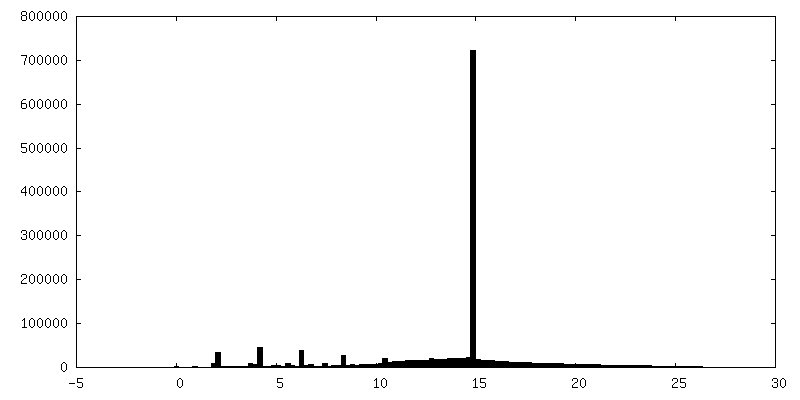
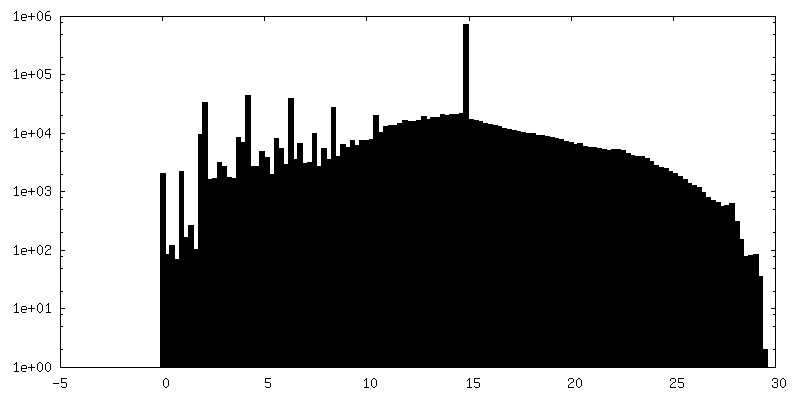
- Sample components
Sample components
-Entire : Influenza A virus
| Entire | Name: Influenza A virus |
|---|---|
| Components |
|
-Supramolecule #1: Influenza A virus
| Supramolecule | Name: Influenza A virus / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Influenza A virus / Strain: A/WSN/33 |
-Macromolecule #1: Nucleoprotein
| Macromolecule | Name: Nucleoprotein / type: protein_or_peptide / ID: 1 / Number of copies: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Influenza A virus (strain A/Wilson-Smith/1933 H1N1) Strain: A/Wilson-Smith/1933 H1N1 |
| Molecular weight | Theoretical: 53.114328 KDa |
| Sequence | String: NATEIRASVG KMIDGIGRFY IQMCTELKLS DYEGRLIQNS LTIERMVLSA FDERRNKYLE EHPSAGKDPK KTGGPIYRRV DGKWRRELI LYDKEEIRRI WRQANNGDDA TAGLTHMMIW HSNLNDATYQ RTRALVRTGM DPRMCSLMQG STLPRRSGAA G AAVKGVGT ...String: NATEIRASVG KMIDGIGRFY IQMCTELKLS DYEGRLIQNS LTIERMVLSA FDERRNKYLE EHPSAGKDPK KTGGPIYRRV DGKWRRELI LYDKEEIRRI WRQANNGDDA TAGLTHMMIW HSNLNDATYQ RTRALVRTGM DPRMCSLMQG STLPRRSGAA G AAVKGVGT MVMELIRMIK RGINDRNFWR GENGRRTRIA YERMCNILKG KFQTAAQRTM VDQVRESRNP GNAEFEDLIF LA RSALILR GSVAHKSCLP ACVYGSAVAS GYDFEREGYS LVGIDPFRLL QNSQVYSLIR PNENPAHKSQ LVWMACHSAA FED LRVSSF IRGTKVVPRG KLSTRGVQIA SNENMETMES STLELRSRYW AIRTRSGGNT NQQRASSGQI SIQPTFSVQR NLPF DRPTI MAAFTGNTEG RTSDMRTEII RLMESARPED VSFQGRGVFE LSDEKATSPI VPSFDMSNEG SYFF UniProtKB: Nucleoprotein |
-Macromolecule #2: Polypeptide loop
| Macromolecule | Name: Polypeptide loop / type: protein_or_peptide / ID: 2 / Number of copies: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Influenza A virus / Strain: A/WSN/33 |
| Molecular weight | Theoretical: 2.107346 KDa |
| Sequence | String: SSGQISIQPT FSVQRNLPF |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | helical reconstruction |
| Aggregation state | helical array |
-Sample preparation
| Buffer | pH: 7.4 / Details: TN buffer (50 mM Tris-HCl, 150 mM KCl) |
|---|---|
| Grid | Model: Quantifoil R2/2 / Material: COPPER/RHODIUM / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 15 sec. / Pretreatment - Atmosphere: AIR |
| Vitrification | Cryogen name: ETHANE / Instrument: LEICA EM CPC |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON II (4k x 4k) / Detector mode: INTEGRATING / Digitization - Frames/image: 3-68 / Number real images: 420 / Average electron dose: 2.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 3.0 µm / Nominal defocus min: 1.5 µm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Final reconstruction | Applied symmetry - Helical parameters - Δz: 33.63 Å Applied symmetry - Helical parameters - Δ&Phi: -65.39 ° Applied symmetry - Helical parameters - Axial symmetry: D1 (2x1 fold dihedral) Algorithm: BACK PROJECTION / Resolution.type: BY AUTHOR / Resolution: 11.0 Å / Resolution method: OTHER / Software - Name: RELION / Details: MonoRes Software Xmipp/SCIPION / Number images used: 2367 |
|---|---|
| Segment selection | Number selected: 137461 / Software - Name: Xmipp / Details: Manual picking |
| Startup model | Type of model: OTHER Details: Initial model created using Iterative Helical Real Space Reconstruction |
| Final angle assignment | Type: NOT APPLICABLE / Software - Name: RELION |
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
| Output model | PDB-6h9g: |

