 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-8562: CryoEM structure of an influenza virus receptor-binding site anti... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8562 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
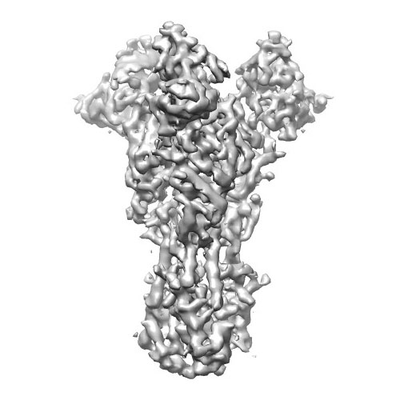
| Title | CryoEM structure of an influenza virus receptor-binding site antibody-antigen interface - Class 2 | |||||||||
 Map data Map data | influenza virus receptor-binding site antibody-antigen interface - Class 2 | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | viral glycoprotein / hemagglutinin / antibody fragment / VIRAL PROTEIN-IMMUNE SYSTEM complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationviral budding from plasma membrane / clathrin-dependent endocytosis of virus by host cell / host cell surface receptor binding / fusion of virus membrane with host plasma membrane / fusion of virus membrane with host endosome membrane / viral envelope / virion attachment to host cell / host cell plasma membrane / virion membrane / membrane Similarity search - Function | |||||||||
| Biological species |  Influenza A virus (A/Solomon Islands/3/2006(H1N1)) / Influenza A virus (A/Solomon Islands/3/2006(H1N1)) /  Influenza A virus / Influenza A virus /  Homo sapiens (human) Homo sapiens (human) | |||||||||
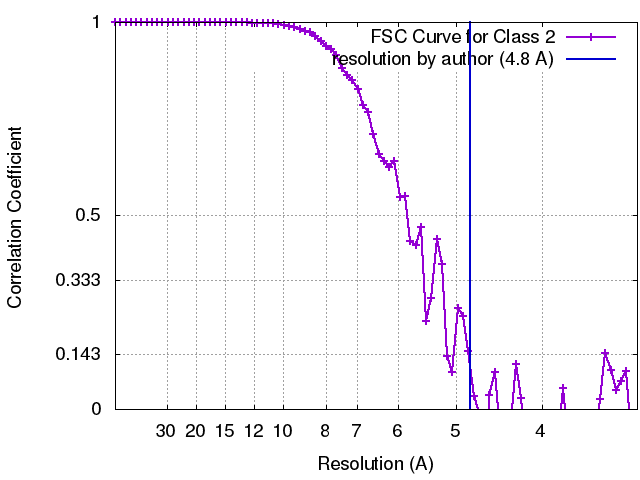
| Method | single particle reconstruction / cryo EM / Resolution: 4.8 Å | |||||||||
 Authors Authors | Liu Y / Pan J | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: J Mol Biol / Year: 2017 Title: CryoEM Structure of an Influenza Virus Receptor-Binding Site Antibody-Antigen Interface. Authors: Yuhang Liu / Junhua Pan / Simon Jenni / Donald D Raymond / Tim Caradonna / Khoi T Do / Aaron G Schmidt / Stephen C Harrison / Nikolaus Grigorieff / Abstract: Structure-based vaccine design depends on extensive structural analyses of antigen-antibody complexes.Single-particle electron cryomicroscopy (cryoEM) can circumvent some of the problems of x-ray ...Structure-based vaccine design depends on extensive structural analyses of antigen-antibody complexes.Single-particle electron cryomicroscopy (cryoEM) can circumvent some of the problems of x-ray crystallography as a pipeline for obtaining the required structures. We have examined the potential of single-particle cryoEM for determining the structure of influenza-virus hemagglutinin (HA):single-chain variable-domain fragment complexes, by studying a complex we failed to crystallize in pursuing an extended project on the human immune response to influenza vaccines.The result shows that a combination of cryoEM and molecular modeling can yield details of the antigen-antibody interface, although small variation in the twist of the rod-likeHA trimer limited the overall resolution to about 4.5Å.Comparison of principal 3D classes suggests ways to modify the HA trimer to overcome this limitation. A closely related antibody from the same donor did yield crystals when bound with the same HA, giving us an independent validation of the cryoEM results.The two structures also augment our understanding of receptor-binding site recognition by antibodies that neutralize a wide range of influenza-virus variants. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8562.map.gz | 1.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8562-v30.xmlemd-8562.xml | 18.8 KB 18.8 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_8562_fsc.xml | 7.6 KB | Display | FSC data file |
| Images |  emd_8562.png emd_8562.png | 59.1 KB | ||
| Filedesc metadata | emd-8562.cif.gz | 7 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8562ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8562 http://ftp.pdbj.org/pub/emdb/structures/EMD-8562ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8562 | HTTPS FTP |
-Related structure data
| Related structure data |  5uk0MC  8561C  8563C  8564C  5ug0C  5ujzC  5uk1C  5uk2C C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |






-Map
| File | Download / File: emd_8562.map.gz / Format: CCP4 / Size: 6.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | influenza virus receptor-binding site antibody-antigen interface - Class 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.64 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
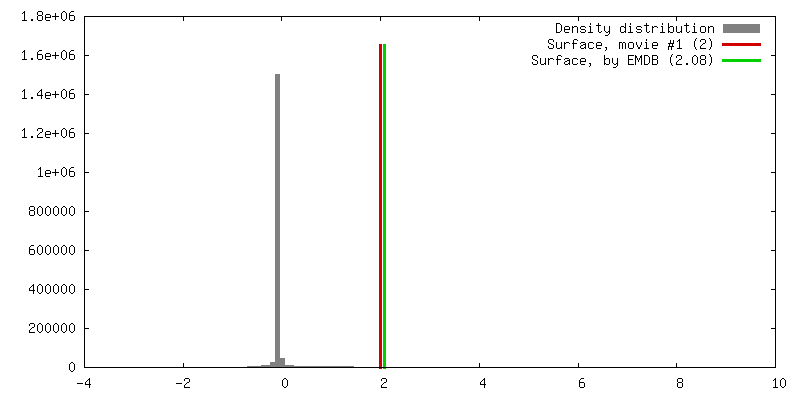
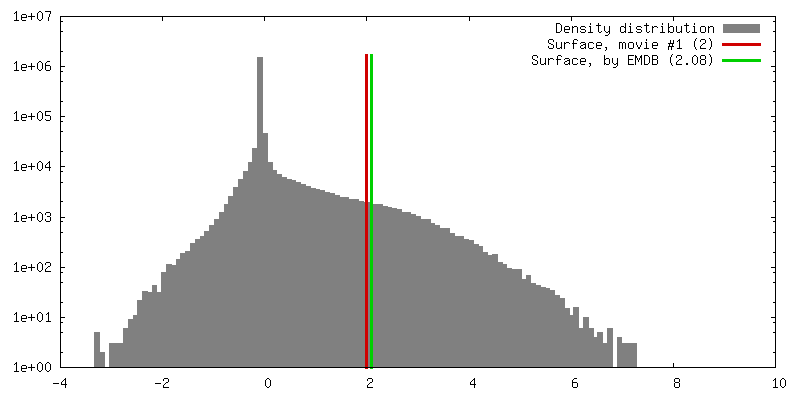
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)

















-Supplemental data
- Sample components
Sample components
-Entire : Influenza-virus hemagglutinin H1 complexed with K1915 single-chai...
| Entire | Name: Influenza-virus hemagglutinin H1 complexed with K1915 single-chain variable-domain fragment |
|---|---|
| Components |
|
-Supramolecule #1: Influenza-virus hemagglutinin H1 complexed with K1915 single-chai...
| Supramolecule | Name: Influenza-virus hemagglutinin H1 complexed with K1915 single-chain variable-domain fragment type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#3 Details: The complex consists of three hemagglutinin head domains bound to three single-chain variable-domain fragments. |
|---|---|
| Source (natural) | Organism: Influenza A virus (A/Solomon Islands/3/2006(H1N1)) |
| Molecular weight | Theoretical: 320 KDa |
-Macromolecule #1: Hemagglutinin HA1
| Macromolecule | Name: Hemagglutinin HA1 / type: protein_or_peptide / ID: 1 / Number of copies: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Influenza A virus (A/Solomon Islands/3/2006(H1N1)) Strain: A/Solomon Islands/3/2006(H1N1) |
| Molecular weight | Theoretical: 36.024344 KDa |
| Recombinant expression | Organism:  Trichoplusia ni (cabbage looper) Trichoplusia ni (cabbage looper) |
| Sequence | String: EDTICIGYHA NNSTDTVDTV LEKNVTVTHS VNLLEDSHNG KLCLLKGIAP LQLGNCSVAG WILGNPECEL LISRESWSYI VEKPNPENG TCYPGHFADY EELREQLSSV SSFERFEIFP KESSWPNHTT TGVSASCSHN GESSFYKNLL WLTGKNGLYP N LSKSYANN ...String: EDTICIGYHA NNSTDTVDTV LEKNVTVTHS VNLLEDSHNG KLCLLKGIAP LQLGNCSVAG WILGNPECEL LISRESWSYI VEKPNPENG TCYPGHFADY EELREQLSSV SSFERFEIFP KESSWPNHTT TGVSASCSHN GESSFYKNLL WLTGKNGLYP N LSKSYANN KEKEVLVLWG VHHPPNIGDQ RALYHTENAY VSVVSSHYSR KFTPEIAKRP KVRDREGRIN YYWTLLEPGD TI IFEANGN LIAPRYAFAL SRGFGSGIIN SNAPMDECDA KCQTPQGAIN SSLPFQNVHP VTIGECPKYV RSAKLRMVTG LRN IPS UniProtKB: Hemagglutinin |
-Macromolecule #2: Hemagglutinin HA2
| Macromolecule | Name: Hemagglutinin HA2 / type: protein_or_peptide / ID: 2 / Number of copies: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Influenza A virus / Strain: A/Solomon Islands/3/2006(H1N1) |
| Molecular weight | Theoretical: 19.841041 KDa |
| Recombinant expression | Organism: Trichoplusia ni (cabbage looper) |
| Sequence | String: GLFGAIAGFI EGGWTGMVDG WYGYHHQNEQ GSGYAADQKS TQNAINGITN KVNSVIEKMN TQFTAVGKEF NKLERRMENL NKKVDDGFI DIWTYNAELL VLLENERTLD FHDSNVKNLY EKVKSQLKNN AKEIGNGCFE FYHKCNDECM ESVKNGTYDY P KYSEESKL NREKI UniProtKB: Hemagglutinin |
-Macromolecule #3: scFV
| Macromolecule | Name: scFV / type: protein_or_peptide / ID: 3 / Number of copies: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 27.629385 KDa |
| Recombinant expression | Organism: Homo sapiens (human) |
| Sequence | String: EIVLTQSPGT LSLSPGERAT LSCRASQSIS GYYLTWYQQK PGQAPRLLIY GASSRATGIP DRFSGSGSGT DFTLTISRLE PEDFAVYYC QQYGISPVIT FGGGTNVEIK GGGGGSGGGG SGGGGSEVQL VESGGGLVQP GGSLRLSCAA SGFTFNIYDM H WVRQAPGK ...String: EIVLTQSPGT LSLSPGERAT LSCRASQSIS GYYLTWYQQK PGQAPRLLIY GASSRATGIP DRFSGSGSGT DFTLTISRLE PEDFAVYYC QQYGISPVIT FGGGTNVEIK GGGGGSGGGG SGGGGSEVQL VESGGGLVQP GGSLRLSCAA SGFTFNIYDM H WVRQAPGK GLEWVSGLTT GGDTSYSGSV RGRFSISREN AKNSLYLQMN NLRAGDTAAY FCVRGVREVG ATGGDPFYYA MA VWGQGTT VTVSSASGSS GSGHHHHHH |
-Macromolecule #6: 2-acetamido-2-deoxy-beta-D-glucopyranose
| Macromolecule | Name: 2-acetamido-2-deoxy-beta-D-glucopyranose / type: ligand / ID: 6 / Number of copies: 18 / Formula: NAG |
|---|---|
| Molecular weight | Theoretical: 221.208 Da |
| Chemical component information |  ChemComp-NAG: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.1 mg/mL |
|---|---|
| Buffer | pH: 8 Details: Beta-octylglucoside was added to a final concentration of 0.07% w/v to induce more variable particle orientations. |
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 90 % / Chamber temperature: 298 K / Instrument: FEI VITROBOT MARK I |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 70.0 K / Max: 70.0 K |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Digitization - Dimensions - Width: 7676 pixel / Digitization - Dimensions - Height: 7420 pixel / Digitization - Frames/image: 1-38 / Number grids imaged: 1 / Number real images: 10281 / Average exposure time: 13.0 sec. / Average electron dose: 40.0 e/Å2 Details: The exposure rate was 8 electrons/physical pixel/second. |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated defocus max: 9.0 µm / Calibrated defocus min: 1.0 µm / Calibrated magnification: 30444 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal magnification: 18000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Details | Domains were initially placed manually using Chimera. The HA trimer was from PDB ID 5UGY. Fab was initially obtained using MODELLER, with PDB ID 4K8R as template. The structure was refined using PHENIX. |
|---|---|
| Refinement | Space: REAL / Protocol: OTHER / Target criteria: Correlation coefficient |
| Output model | PDB-5uk0: |
