 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-50566: Cryo-EM structure of S. cerevisiae Rai1-Rat1-Rtt103(252-273) complex. -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of S. cerevisiae Rai1-Rat1-Rtt103(252-273) complex. | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Rai1 nuclease / Rtt103 binding / structured / RNA binding / RNA BINDING PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationRNA polymerase II termination complex / sno(s)RNA processing / positive regulation of termination of RNA polymerase II transcription / RNA NAD+-cap (NAD+-forming) hydrolase activity / termination of RNA polymerase II transcription, poly(A)-coupled / Las1 complex / termination of RNA polymerase II transcription, exosome-dependent / Butyrate Response Factor 1 (BRF1) binds and destabilizes mRNA / Tristetraprolin (TTP, ZFP36) binds and destabilizes mRNA / phosphodiesterase decapping endonuclease activity ...RNA polymerase II termination complex / sno(s)RNA processing / positive regulation of termination of RNA polymerase II transcription / RNA NAD+-cap (NAD+-forming) hydrolase activity / termination of RNA polymerase II transcription, poly(A)-coupled / Las1 complex / termination of RNA polymerase II transcription, exosome-dependent / Butyrate Response Factor 1 (BRF1) binds and destabilizes mRNA / Tristetraprolin (TTP, ZFP36) binds and destabilizes mRNA / phosphodiesterase decapping endonuclease activity / deadenylation-independent decapping of nuclear-transcribed mRNA / mRNA 5'-diphosphatase activity / RNA polymerase II C-terminal domain phosphoserine binding / nuclear polyadenylation-dependent rRNA catabolic process / NAD-cap decapping / 5'-3' RNA exonuclease activity / nuclear mRNA surveillance / transposable element silencing / mRNA 3'-end processing / maturation of 5.8S rRNA from tricistronic rRNA transcript (SSU-rRNA, 5.8S rRNA, LSU-rRNA) / RNA polymerase II transcribes snRNA genes / cleavage in ITS2 between 5.8S rRNA and LSU-rRNA of tricistronic rRNA transcript (SSU-rRNA, 5.8S rRNA, LSU-rRNA) / RNA polymerase II complex binding / negative regulation of transcription elongation by RNA polymerase II / nuclear-transcribed mRNA catabolic process / Hydrolases; Acting on acid anhydrides; In phosphorus-containing anhydrides / enzyme regulator activity / maturation of LSU-rRNA from tricistronic rRNA transcript (SSU-rRNA, 5.8S rRNA, LSU-rRNA) / mRNA splicing, via spliceosome / mRNA processing / rRNA processing / site of double-strand break / Hydrolases; Acting on ester bonds; Exoribonucleases producing 5'-phosphomonoesters / rRNA binding / nucleotide binding / mRNA binding / chromatin / mitochondrion / DNA binding / RNA binding / metal ion binding / nucleus / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.65 Å | |||||||||
 Authors Authors | Dikunova A / Kubicek K / Noskova N / Stefl R | |||||||||
| Funding support |  Czech Republic, 1 items Czech Republic, 1 items
| |||||||||
 Citation Citation | Journal: Structure / Year: 2025 Title: Assembly of the Xrn2/Rat1-Rai1-Rtt103 termination complexes in mesophilic and thermophilic organisms. Authors: Alzbeta Dikunova / Nikola Noskova / Jan H Overbeck / Martin Polak / David Stelzig / David Zapletal / Karel Kubicek / Jiri Novacek / Remco Sprangers / Richard Stefl /  Abstract: The 5'-3' exoribonuclease Xrn2, known as Rat1 in yeasts, terminates mRNA transcription by RNA polymerase II (RNAPII). In the torpedo model of termination, the activity of Xrn2/Rat1 is enhanced by ...The 5'-3' exoribonuclease Xrn2, known as Rat1 in yeasts, terminates mRNA transcription by RNA polymerase II (RNAPII). In the torpedo model of termination, the activity of Xrn2/Rat1 is enhanced by Rai1, which is recruited to the termination site by Rtt103, an adaptor protein binding to the RNAPII C-terminal domain (CTD). The overall architecture of the Xrn2/Rat1-Rai1-Rtt103 complex remains unknown. We combined structural biology methods to characterize the torpedo complex from Saccharomyces cerevisiae and Chaetomium thermophilum. Comparison of the structures from these organisms revealed a conserved protein core fold of the subunits, but significant variability in their interaction interfaces. We found that in the mesophile, Rtt103 utilizes an unstructured region to augment a Rai1 β-sheet, while in the thermophile Rtt103 binds to a C-terminal helix of Rai1 via its CTD-interacting domain with an α-helical fold. These different torpedo complex assemblies reflect adaptations to the environment and impact complex recruitment to RNAPII. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_50566.map.gz | 108.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-50566-v30.xmlemd-50566.xml | 22.3 KB 22.3 KB | Display Display | EMDB header |
| Images |  emd_50566.png emd_50566.png | 70.6 KB | ||
| Filedesc metadata | emd-50566.cif.gz | 7.6 KB | ||
| Others | emd_50566_half_map_1.map.gzemd_50566_half_map_2.map.gz | 200.2 MB 200.2 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-50566ftp://ftp.pdbj.org/pub/emdb/structures/EMD-50566 http://ftp.pdbj.org/pub/emdb/structures/EMD-50566ftp://ftp.pdbj.org/pub/emdb/structures/EMD-50566 | HTTPS FTP |
-Validation report
| Summary document | emd_50566_validation.pdf.gz | 1018.9 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_50566_full_validation.pdf.gz | 1018.5 KB | Display | |
| Data in XML | emd_50566_validation.xml.gz | 15.6 KB | Display | |
| Data in CIF | emd_50566_validation.cif.gz | 18.5 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-50566ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-50566 | HTTPS FTP |
-Related structure data
| Related structure data |  9fmsMC  8q6vC  9exsC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |


-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |




-Map
| File | Download / File: emd_50566.map.gz / Format: CCP4 / Size: 216 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.97734 Å | ||||||||||||||||||||||||||||||||||||
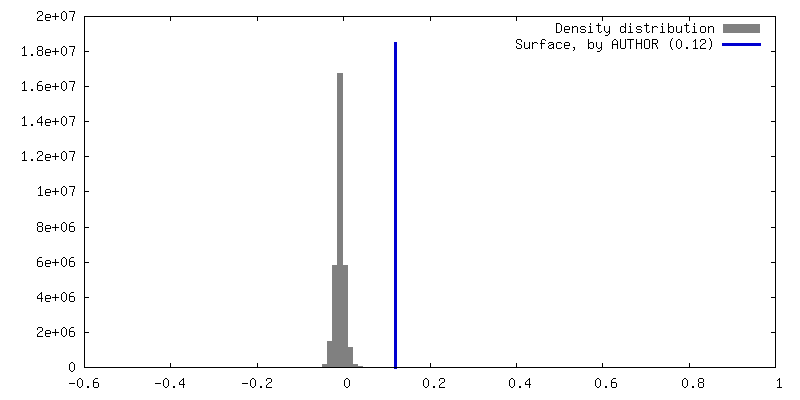
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Half map: #2
| File | emd_50566_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #1
| File | emd_50566_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |







- Sample components
Sample components
-Entire : Cryo-EM structure of S. cerevisiae Rai1-Rat1-Rtt103(252-273) complex.
| Entire | Name: Cryo-EM structure of S. cerevisiae Rai1-Rat1-Rtt103(252-273) complex. |
|---|---|
| Components |
|
-Supramolecule #1: Cryo-EM structure of S. cerevisiae Rai1-Rat1-Rtt103(252-273) complex.
| Supramolecule | Name: Cryo-EM structure of S. cerevisiae Rai1-Rat1-Rtt103(252-273) complex. type: complex / ID: 1 / Parent: 0 / Macromolecule list: #3, #1-#2 |
|---|---|
| Source (natural) | Organism: |
-Macromolecule #1: 5'-3' exoribonuclease 2
| Macromolecule | Name: 5'-3' exoribonuclease 2 / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 116.08493 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: MGVPSFFRWL SRKYPKIISP VLEEQPQIVD GVILPLDYSA SNPNGELDNL YLDMNGIVHP CSHPENKPPP ETEDEMLLAV FEYTNRVLN MARPRKVLVM AVDGVAPRAK MNQQRARRFR SARDAQIENE AREEIMRQRE EVGEIIDDAV RNKKTWDSNA I TPGTPFMD ...String: MGVPSFFRWL SRKYPKIISP VLEEQPQIVD GVILPLDYSA SNPNGELDNL YLDMNGIVHP CSHPENKPPP ETEDEMLLAV FEYTNRVLN MARPRKVLVM AVDGVAPRAK MNQQRARRFR SARDAQIENE AREEIMRQRE EVGEIIDDAV RNKKTWDSNA I TPGTPFMD KLAAALRYWT AFKLATDPGW KNLQVIISDA TVPGEGEHKI MNFIRSQRAD PEYNPNTTHC IYGLDADLIF LG LATHEPH FKILREDVFA QDNRKRNNLK DTINMTEEEK QFLQKQNSEQ PFLWLHINVL REYLSAELWV PGLPFTFDLE RAI DDWVFM CFFCGNDFLP HLPCLDVREN SIDILLDIWK VVLPKLKTYM TCDGVLNLPS VETLLQHLGS REGDIFKTRH IQEA RKKEA FERRKAQKNM SKGQDRHPTV ATEQLQMYDT QGNLAKGSWN LTTSDMVRLK KELMLANEGN EEAIAKVKQQ SDKNN ELMK DISKEEIDDA VSKANKTNFN LAEVMKQKII NKKHRLEKDN EEEEIAKDSK KVKTEKAESE CDLDAEIKDE IVADVN DRE NSETTEVSRD SPVHSTVNVS EGPKNGVFDT DEFVKLFEPG YHERYYTAKF HVTPQDIEQL RKDMVKCYIE GVAWVLM YY YQGCASWNWF YPYHYAPLAT DFHGFSHLEI KFEEGTPFLP YEQLMSVLPA ASGHALPKIF RSLMSEPDSE IIDFYPEE F PIDMNGKKMS WQGIALLPFI DQDRLLTAVR AQYPLLSDAE RARNIRGEPV LLISNKNANY ERFSKKLYSK ENNNNNVVV KFQHFKSGLS GIVSKDVEGF ELNGKIVCPI QGGSLPNLST TLILKMSYRL IPLPSRNKSI ILNGFIPSEP VLTAYDLDSI MYKYNNQNY SRRWNFGNDL KQNIVPVGPK GITQYKPRTG GYRAFFYFAE LSRNNVQPAH NYGRNSYNSQ PGFNNSRYDG G NNNYRQNS NYRNNNYSGN RNSGQYSGNS YSRNNKQSRY DNSRANRR UniProtKB: 5'-3' exoribonuclease 2 |
-Macromolecule #2: Regulator of Ty1 transposition protein 103
| Macromolecule | Name: Regulator of Ty1 transposition protein 103 / type: protein_or_peptide / ID: 2 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 2.454404 KDa |
| Sequence | String: KNVDEDNIIP TYEVGDGDDD DD UniProtKB: Regulator of Ty1 transposition protein 103 |
-Macromolecule #3: Decapping nuclease RAI1
| Macromolecule | Name: Decapping nuclease RAI1 / type: protein_or_peptide / ID: 3 / Number of copies: 1 / Enantiomer: LEVO EC number: Hydrolases; Acting on acid anhydrides; In phosphorus-containing anhydrides |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 44.571445 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MGVSANLFVK QRGSTTALKQ PKEIGFYSRT KDEEYLISDD TNLNYYYLPD AELDRKLDLS SGFQKFKDYY KDFEDRCSLR GLLETIESS ERHKGKKINA DIITFRGIAR KLISCAFDSP SFNTVDLRIV SFNGQLFIKE VPEAVNAAKA SSATEAGRNI N QDLNVFTG ...String: MGVSANLFVK QRGSTTALKQ PKEIGFYSRT KDEEYLISDD TNLNYYYLPD AELDRKLDLS SGFQKFKDYY KDFEDRCSLR GLLETIESS ERHKGKKINA DIITFRGIAR KLISCAFDSP SFNTVDLRIV SFNGQLFIKE VPEAVNAAKA SSATEAGRNI N QDLNVFTG YKFETLATLS NPLQYTPREV IEKRTKRIVS HGDEYISVVR TGVGNCKLIL GAEVDCIFDF KENGRDNLKH YA ELKCTQQ VANISDTHKF ERKLFRTWLQ CFLVGIPRII YGFKDDHYVL KTVEEFSTEE VPVLLKNNNP QVGSACLEAI KWY GLLTEW LLKMIPRDED PHSQIRAFKL VFENNHLRLS EIEESDEEYS GLIDGEHILS NGFKEWRKSL K UniProtKB: Decapping nuclease RAI1 |
-Macromolecule #4: MAGNESIUM ION
| Macromolecule | Name: MAGNESIUM ION / type: ligand / ID: 4 / Number of copies: 1 / Formula: MG |
|---|---|
| Molecular weight | Theoretical: 24.305 Da |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.3 mg/mL | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 Component:
| |||||||||||||||
| Grid | Model: UltrAuFoil R1.2/1.3 / Material: GOLD / Support film - Material: GOLD / Support film - topology: CONTINUOUS / Pretreatment - Type: GLOW DISCHARGE | |||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277.15 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Average electron dose: 40.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 3.5 µm / Nominal defocus min: 0.8 µm |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Initial model |
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Details | As initial model was used previously estimated ScRai1-Rat1 dimer. The position of investigated peptide was predicted by AlphaFold and manually built into the corresponding cryo-EM using Coot. For final geometry estimation was used ISOLDE. | ||||||||
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT | ||||||||
| Output model | PDB-9fms: |
