National Institutes of Health/National Institute of General Medical Sciences
R35GM128674
United States
National Institutes of Health/National Institute Of Allergy and Infectious Diseases
AI116566
United States
National Institutes of Health/National Institute Of Allergy and Infectious Diseases
AI118863
United States
Citation
Journal: PLoS Genet / Year: 2019 Title: PilT and PilU are homohexameric ATPases that coordinate to retract type IVa pili. Authors: Jennifer L Chlebek / Hannah Q Hughes / Aleksandra S Ratkiewicz / Rasman Rayyan / Joseph Che-Yen Wang / Brittany E Herrin / Triana N Dalia / Nicolas Biais / Ankur B Dalia / Abstract: Bacterial type IV pili are critical for diverse biological processes including horizontal gene transfer, surface sensing, biofilm formation, adherence, motility, and virulence. These dynamic ...Bacterial type IV pili are critical for diverse biological processes including horizontal gene transfer, surface sensing, biofilm formation, adherence, motility, and virulence. These dynamic appendages extend and retract from the cell surface. In many type IVa pilus systems, extension occurs through the action of an extension ATPase, often called PilB, while optimal retraction requires the action of a retraction ATPase, PilT. Many type IVa systems also encode a homolog of PilT called PilU. However, the function of this protein has remained unclear because pilU mutants exhibit inconsistent phenotypes among type IV pilus systems and because it is relatively understudied compared to PilT. Here, we study the type IVa competence pilus of Vibrio cholerae as a model system to define the role of PilU. We show that the ATPase activity of PilU is critical for pilus retraction in PilT Walker A and/or Walker B mutants. PilU does not, however, contribute to pilus retraction in ΔpilT strains. Thus, these data suggest that PilU is a bona fide retraction ATPase that supports pilus retraction in a PilT-dependent manner. We also found that a ΔpilU mutant exhibited a reduction in the force of retraction suggesting that PilU is important for generating maximal retraction forces. Additional in vitro and in vivo data show that PilT and PilU act as independent homo-hexamers that may form a complex to facilitate pilus retraction. Finally, we demonstrate that the role of PilU as a PilT-dependent retraction ATPase is conserved in Acinetobacter baylyi, suggesting that the role of PilU described here may be broadly applicable to other type IVa pilus systems.
History
Deposition
Sep 24, 2019
-
Header (metadata) release
Oct 23, 2019
-
Map release
Oct 30, 2019
-
Update
Oct 30, 2019
-
Current status
Oct 30, 2019
Processing site: RCSB / Status: Released
-
Structure visualization
Movie
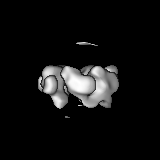
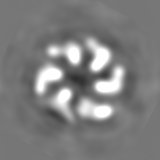
Surface view with section colored by density value
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample
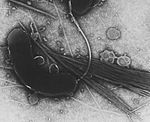 Vibrio cholerae (bacteria)
Vibrio cholerae (bacteria) Authors
Authors United States, 3 items
United States, 3 items  Citation
Citation Structure visualization
Structure visualization Movie viewer
Movie viewer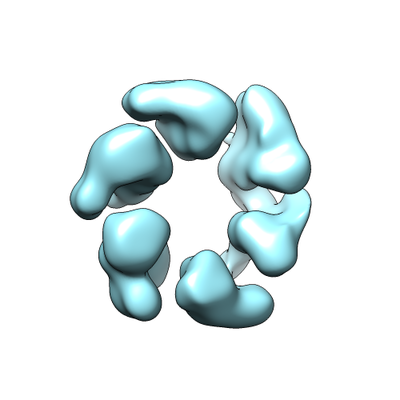
 Downloads & links
Downloads & links emd_20757.png
emd_20757.png http://ftp.pdbj.org/pub/emdb/structures/EMD-20757
http://ftp.pdbj.org/pub/emdb/structures/EMD-20757





 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)
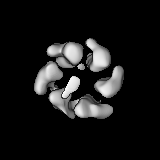
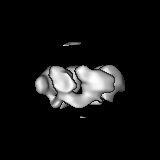















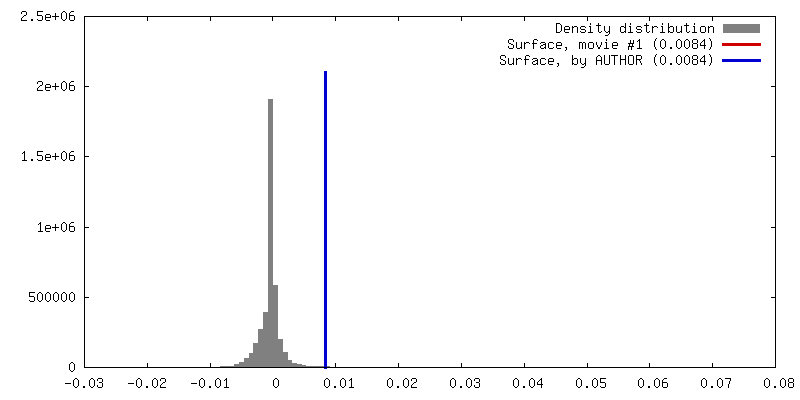
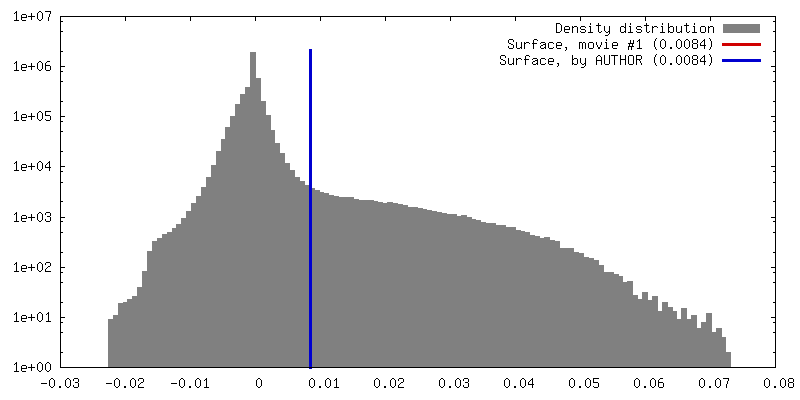
 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN