Bacterial/eukaryotic lysine-tRNA ligase, class II / Lysine-tRNA ligase, class II, N-terminal / Lysine-tRNA ligase, class II / Lysyl-tRNA synthetase, class II, C-terminal / Aminoacyl-tRNA synthetase, class II (D/K/N) / tRNA synthetases class II (D, K and N) / OB-fold nucleic acid binding domain, AA-tRNA synthetase-type / OB-fold nucleic acid binding domain / Bira Bifunctional Protein; Domain 2 / BirA Bifunctional Protein; domain 2 ...Bacterial/eukaryotic lysine-tRNA ligase, class II / Lysine-tRNA ligase, class II, N-terminal / Lysine-tRNA ligase, class II / Lysyl-tRNA synthetase, class II, C-terminal / Aminoacyl-tRNA synthetase, class II (D/K/N) / tRNA synthetases class II (D, K and N) / OB-fold nucleic acid binding domain, AA-tRNA synthetase-type / OB-fold nucleic acid binding domain / Bira Bifunctional Protein; Domain 2 / BirA Bifunctional Protein; domain 2 / Aminoacyl-tRNA synthetase, class II / Aminoacyl-transfer RNA synthetases class-II family profile. / Class II Aminoacyl-tRNA synthetase/Biotinyl protein ligase (BPL) and lipoyl protein ligase (LPL) / Nucleic acid-binding proteins / OB fold (Dihydrolipoamide Acetyltransferase, E2P) / Nucleic acid-binding, OB-fold / Beta Barrel / 2-Layer Sandwich / Mainly Beta / Alpha Beta Similarity search - Domain/homology
Resolution: 3.08→42.17 Å / Cor.coef. Fo:Fc: 0.922 / Cor.coef. Fo:Fc free: 0.887 / SU B: 72.432 / SU ML: 0.554 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.537 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.298
1141
4.8 %
RANDOM
Rwork
0.243
-
-
-
obs
0.246
22625
99.7 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information lysine-tRNA ligase /
lysine-tRNA ligase / 
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads













 PDBj
PDBj

 Assembly
Assembly

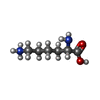
 Type: L-peptide linking / Mass: 147.195 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H15N2O2
Type: L-peptide linking / Mass: 147.195 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H15N2O2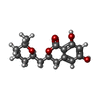
 Mass: 292.327 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H20O5
Mass: 292.327 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H20O5
 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Sample preparation
Sample preparation / Beamline: I03 / Wavelength: 0.9763 Å
/ Beamline: I03 / Wavelength: 0.9763 Å Processing
Processing