 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4zcm: Crystal Structure of Escherichia coli GTPase BipA/TypA Complexed ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4zcm | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Escherichia coli GTPase BipA/TypA Complexed with ppGpp | ||||||
 Components Components | GTP-binding protein TypA/BipA | ||||||
 Keywords Keywords | GTP-binding protein / BipA / GTPase / Nucleotide | ||||||
| Function / homology |  Function and homology information Function and homology informationguanosine tetraphosphate binding / protein folding chaperone / response to cold / ribosome binding / response to heat / ribosomal large subunit assembly / Hydrolases; Acting on acid anhydrides; Acting on GTP to facilitate cellular and subcellular movement / tRNA binding / rRNA binding / translation ...guanosine tetraphosphate binding / protein folding chaperone / response to cold / ribosome binding / response to heat / ribosomal large subunit assembly / Hydrolases; Acting on acid anhydrides; Acting on GTP to facilitate cellular and subcellular movement / tRNA binding / rRNA binding / translation / ribonucleoprotein complex / GTPase activity / GTP binding / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.31 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.31 Å | ||||||
 Authors Authors | Fan, H.T. / Hahm, J. / Diggs, S. / Blaha, G. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2015 Title: Structural and Functional Analysis of BipA, a Regulator of Virulence in Enteropathogenic Escherichia coli. Authors: Fan, H. / Hahm, J. / Diggs, S. / Perry, J.J. / Blaha, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4zcm.cif.gz | 452.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4zcm.ent.gz | 370 KB | Display | PDB format |
| PDBx/mmJSON format | 4zcm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zc/4zcmftp://data.pdbj.org/pub/pdb/validation_reports/zc/4zcm | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4zciSC  4zckC  4zclC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 70992.289 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Strain: K12 / Gene: typA, bipA, yihK, b3871, JW5571 / Production host: #2: Chemical | ChemComp-NCO / 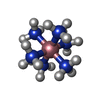  Mass: 161.116 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: CoH18N6 Mass: 161.116 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: CoH18N6#3: Chemical | 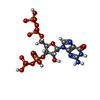  Type: RNA linking / Mass: 603.160 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N5O17P4 Type: RNA linking / Mass: 603.160 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N5O17P4#4: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.85 Å3/Da / Density % sol: 56.79 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 8 Details: 100 mM Tris-HCl, 2 % PEG 6000, and 5 mM [Co(NH3)6]Cl3, pH 8.0 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 5.0.1 / Wavelength: 0.98 Å / Beamline: 5.0.1 / Wavelength: 0.98 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Dec 20, 2014 |
| Radiation | Monochromator: Single crystal, cylindrically bent, Si(220) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 |
| Reflection | Resolution: 3.31→48.471 Å / Num. obs: 23655 / % possible obs: 98.9 % / Redundancy: 3 % / Biso Wilson estimate: 70.4 Å2 / Rmerge(I) obs: 0.17 / Net I/σ(I): 7 |
| Reflection shell | Resolution: 3.31→3.58 Å / Redundancy: 2.9 % / Rmerge(I) obs: 0.7 / Mean I/σ(I) obs: 1.8 / % possible all: 98.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4ZCI Resolution: 3.31→48.471 Å / SU ML: 0.48 / Cross valid method: FREE R-VALUE / σ(F): 1.36 / Phase error: 33.69 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 104 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.31→48.471 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
