 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 465d | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF THE TOPOISOMERASE II POISON BOUND TO DNA | ||||||||||||||||||
 Components Components | DNA (5'-D(* Keywords KeywordsDNA / TOPOISOMERASE II POISON / HEXANUCLEOTIDE / D(CGTACG)2 / 9-AMINO-DACA | Function / homology | Chem-9AD / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / ISOMORPHOUS REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / ISOMORPHOUS REPLACEMENT / Resolution: 1.6 Å  Authors AuthorsAdams, A. / Guss, J.M. / Collyer, C.A. / Denny, W.A. / Wakelin, L.P. |  CitationJournal: Biochemistry / Year: 1999 CitationJournal: Biochemistry / Year: 1999Title: Crystal structure of the topoisomerase II poison 9-amino-[N-(2-dimethylamino)ethyl]acridine-4-carboxamide bound to the DNA hexanucleotide d(CGTACG)2. Authors: Adams, A. / Guss, J.M. / Collyer, C.A. / Denny, W.A. / Wakelin, L.P. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 465d.cif.gz | 15.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb465d.ent.gz | 8.8 KB | Display | PDB format |
| PDBx/mmJSON format | 465d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 465d_validation.pdf.gz | 436.6 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 465d_full_validation.pdf.gz | 441.6 KB | Display | |
| Data in XML | 465d_validation.xml.gz | 2.4 KB | Display | |
| Data in CIF | 465d_validation.cif.gz | 3.1 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/65/465dftp://data.pdbj.org/pub/pdb/validation_reports/65/465d | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|



-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 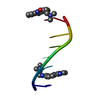
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 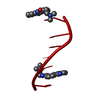
| ||||||||||
| Unit cell |
| ||||||||||
| Components on special symmetry positions |
|
-Components
| #1: DNA chain | Mass: 1809.217 Da / Num. of mol.: 1 / Source method: obtained synthetically | ||
|---|---|---|---|
| #2: Chemical | 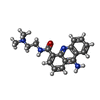  Mass: 308.378 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H20N4O Mass: 308.378 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H20N4O#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 29 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 29 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.76 Å3/Da / Density % sol: 55.41 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 285 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: 0.5 MM DNA, 20 MM SODIUM CACODYLATE BUFFER, PH 6.5, 4 MM MAGNESIUM ACETATE, 1 MM COBALT(II) CHLORIDE, 0.33 MM SPERMINE.4HCL, 1 MM 9-AMINO-[N-(2- DIMETHYLAMINO)ETHYL]ACRIDINE-4-CARBOXAMIDE ...Details: 0.5 MM DNA, 20 MM SODIUM CACODYLATE BUFFER, PH 6.5, 4 MM MAGNESIUM ACETATE, 1 MM COBALT(II) CHLORIDE, 0.33 MM SPERMINE.4HCL, 1 MM 9-AMINO-[N-(2- DIMETHYLAMINO)ETHYL]ACRIDINE-4-CARBOXAMIDE AND 9% MPD EQUILIBRATED AGAINST 1 ML OF A SOLUTION OF 45% MPD IN WATER, VAPOR DIFFUSION, SITTING DROP, temperature 285K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 285 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE / Date: Dec 12, 1997 / Details: FOCUSING MIRROR |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→30 Å / Num. all: 2763 / Num. obs: 2763 / % possible obs: 98.4 % / Observed criterion σ(I): 0 / Redundancy: 24.7 % / Rmerge(I) obs: 0.029 / Net I/σ(I): 56.8 |
| Reflection shell | Resolution: 1.6→1.66 Å / Redundancy: 5.3 % / Rmerge(I) obs: 0.142 / Mean I/σ(I) obs: 5 / % possible all: 97 |
| Reflection | *PLUS Num. measured all: 67055 |
| Reflection shell | *PLUS % possible obs: 97 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: ISOMORPHOUS REPLACEMENT Starting model: DDF073 Resolution: 1.6→16 Å / Num. parameters: 791 / Num. restraintsaints: 999 / Cross valid method: THROUGHOUT / σ(F): 0
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER,J.MOL.BIOL.91(1973)201-228 | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 1 / Occupancy sum non hydrogen: 182.5 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→16 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.6 Å / σ(F): 0 / % reflection Rfree: 8.3 % / Rfactor obs: 0.193 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: s_plane_restr / Dev ideal: 0.023 |
