Mass: 18.015 Da / Num. of mol.: 176 / Source method: isolated from a natural source / Formula: H2O
Compound details
ENGINEERED RESIDUE IN CHAIN A, ASN 51 TO ALA ENGINEERED RESIDUE IN CHAIN A, ILE 53 TO SER ...ENGINEERED RESIDUE IN CHAIN A, ASN 51 TO ALA ENGINEERED RESIDUE IN CHAIN A, ILE 53 TO SER ENGINEERED RESIDUE IN CHAIN A, GLU 222 TO GLY
Has protein modification
N
Sequence details
32 RESIDUES DELETED AT THE C-TERMINUS
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.48 Å3/Da / Density % sol: 50 % / Description: NONE
Resolution: 2→35 Å / Cor.coef. Fo:Fc: 0.947 / Cor.coef. Fo:Fc free: 0.92 / SU B: 8.351 / SU ML: 0.106 / Cross valid method: THROUGHOUT / ESU R: 0.176 / ESU R Free: 0.16 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.23005
1098
5.1 %
RANDOM
Rwork
0.18565
-
-
-
obs
0.18788
20305
99.98 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 CAMPYLOBACTER JEJUNI (Campylobacter)
CAMPYLOBACTER JEJUNI (Campylobacter) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads













 PDBj
PDBj Assembly
Assembly


 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2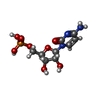
 Mass: 323.197 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N3O8P
Mass: 323.197 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N3O8P Mass: 18.015 Da / Num. of mol.: 176 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 176 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing