 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2g0v | ||||||
|---|---|---|---|---|---|---|---|
| Title | Photolyzed CO L29F Myoglobin: 100ps | ||||||
 Components Components | Myoglobin | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / Time-resolved crystallography / myoglobin / difference refinement / structure-function relationship / intermediate states | ||||||
| Function / homology |  Function and homology information Function and homology informationOxidoreductases; Acting on other nitrogenous compounds as donors / nitrite reductase activity / sarcoplasm / Oxidoreductases; Acting on a peroxide as acceptor; Peroxidases / removal of superoxide radicals / oxygen carrier activity / peroxidase activity / oxygen binding / heme binding / extracellular exosome / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.95 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.95 Å | ||||||
 Authors Authors | Aranda, R. / Levin, E.J. / Schotte, F. / Anfinrud, P.A. / Phillips Jr., G.N. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2006 Title: Time-dependent atomic coordinates for the dissociation of carbon monoxide from myoglobin. Authors: Aranda, R. / Levin, E.J. / Schotte, F. / Anfinrud, P.A. / Phillips Jr., G.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2g0v.cif.gz | 81.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2g0v.ent.gz | 61.2 KB | Display | PDB format |
| PDBx/mmJSON format | 2g0v.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g0/2g0vftp://data.pdbj.org/pub/pdb/validation_reports/g0/2g0v | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2g0rC  2g0sC  2g0xC  2g0zC  2g10C  2g11C  2g12C  2g14C  2splS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Number of models | 2 |
-Components
| #1: Protein | Mass: 17399.180 Da / Num. of mol.: 1 / Mutation: L29F Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|---|
| #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #3: Chemical | ChemComp-HEM /   Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 |
| #4: Chemical | ChemComp-CMO / 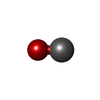  Mass: 28.010 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO Mass: 28.010 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 147 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 147 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.16 Å3/Da / Density % sol: 61.13 % |
|---|---|
| Crystal grow | Temperature: 283 K / pH: 9 Details: 2.6M Ammonium Sulfate, 20mM TrisHCl, 1mM EDTA, pH 9.0, VAPOR DIFFUSION, HANGING DROP, temperature 283K, pH 9.00 |
-Data collection
| Diffraction | Mean temperature: 283 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID09 / Wavelength: 0.79 / Beamline: ID09 / Wavelength: 0.79 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Apr 1, 2002 |
| Radiation | Protocol: LAUE / Monochromatic (M) / Laue (L): L / Scattering type: neutron |
| Radiation wavelength | Wavelength: 0.79 Å / Relative weight: 1 |
| Reflection | Resolution: 1.801→29.884 Å / Num. obs: 18402 / Biso Wilson estimate: 16.6 Å2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY: 2SPL Resolution: 1.95→12.7 Å / Rfactor Rfree error: 0.002 / Data cutoff high absF: 1541641.75 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber / Details: DIFFERENCE REFINEMENT (TERWILLIGER 1995)
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 20.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.95→12.7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.95→2.07 Å / Rfactor Rfree error: 0.011 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
