 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1eeh | ||||||
|---|---|---|---|---|---|---|---|
| Title | UDP-N-ACETYLMURAMOYL-L-ALANINE:D-GLUTAMATE LIGASE | ||||||
 Components Components | UDP-N-ACETYLMURAMOYL-L-ALANINE:D-GLUTAMATE LIGASE | ||||||
 Keywords Keywords |  LIGASE / PEPTIDOGLYCAN SYNTHESIS / MURD / ADP-FORMING ENZYME LIGASE / PEPTIDOGLYCAN SYNTHESIS / MURD / ADP-FORMING ENZYME | ||||||
| Function / homology |  Function and homology informationUDP-N-acetylmuramoyl-L-alanine-D-glutamate ligase / UDP-N-acetylmuramoylalanine-D-glutamate ligase activity / peptidoglycan biosynthetic process / cell wall organization / regulation of cell shape / cell cycle / cell division / ATP binding / identical protein binding / cytoplasm Function and homology informationUDP-N-acetylmuramoyl-L-alanine-D-glutamate ligase / UDP-N-acetylmuramoylalanine-D-glutamate ligase activity / peptidoglycan biosynthetic process / cell wall organization / regulation of cell shape / cell cycle / cell division / ATP binding / identical protein binding / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Escherichia coli (E. coli) Escherichia coli (E. coli) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.9 Å | ||||||
 Authors Authors | Bertrand, J.A. / Fanchon, E. / Martin, L. / Chantalat, L. / Auger, G. / Blanot, D. / van Heijenoort, J. / Dideberg, O. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2000 Title: "Open" structures of MurD: domain movements and structural similarities with folylpolyglutamate synthetase. Authors: Bertrand, J.A. / Fanchon, E. / Martin, L. / Chantalat, L. / Auger, G. / Blanot, D. / van Heijenoort, J. / Dideberg, O. #1: Journal: Embo J. / Year: 1997Title: Crystal Structure of UDP-N-acetylmuramoyl-L-alanine: D-glutamate ligase from Escherichia coli Authors: Bertrand, J.A. / Auger, G. / Fanchon, E. / Martin, L. / Blanot, D. / van Heijenoort, J. / Dideberg, O. #2: Journal: J.Mol.Biol. / Year: 1999Title: Determination of the MurD mechanism through crystallographic analysis of enzyme complexes Authors: Bertrand, J.A. / Auger, G. / Martin, L. / Fanchon, E. / Blanot, D. / Le Beller, D. / van Heijenoort, J. / Dideberg, O. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1eeh.cif.gz | 98.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1eeh.ent.gz | 74.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1eeh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ee/1eehftp://data.pdbj.org/pub/pdb/validation_reports/ee/1eeh | HTTPS FTP |
|---|
-Related structure data















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 46889.250 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Escherichia coli (E. coli) / Production host: Escherichia coli (E. coli)References: UniProt: P14900, UDP-N-acetylmuramoyl-L-alanine-D-glutamate ligase |
|---|---|
| #2: Chemical | ChemComp-UMA / 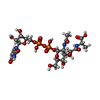  Type: L-peptide linking / Mass: 750.494 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H36N4O20P2 Type: L-peptide linking / Mass: 750.494 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H36N4O20P2 |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 186 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 186 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.28 Å3/Da / Density % sol: 46.06 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 288 K / Method: vapor diffusion, hanging drop / pH: 5.2 Details: PROTEIN SOLUTION: 9.8 mg/ml MURD, 1 mM UMA, 1 mM Sodium Azide, 1 mM DTT, 20 mM HEPES pH 7.5 RESEVOIR: 5-9 % PEG 3350 Monodisperse, 100 mM sodium Acetate pH 5.2, VAPOR DIFFUSION, HANGING DROP, temperature 288K | ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 15 ℃ / pH: 7.5 | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM02 / Wavelength: 1.073 / Beamline: BM02 / Wavelength: 1.073 |
| Detector | Type: XRII-CCD / Detector: CCD / Date: Sep 1, 1997 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.073 Å / Relative weight: 1 |
| Reflection | Resolution: 1.878→13.392 Å / Num. all: 31255 / Num. obs: 31255 / % possible obs: 87.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2.5 % / Biso Wilson estimate: 11.4 Å2 / Rsym value: 0.04 / Net I/σ(I): 13 |
| Reflection shell | Resolution: 1.88→1.94 Å / Redundancy: 1.5 % / Mean I/σ(I) obs: 3.3 / Num. unique all: 1675 / Rsym value: 0.279 / % possible all: 49.1 |
| Reflection | *PLUS Rmerge(I) obs: 0.04 |
| Reflection shell | *PLUS Lowest resolution: 1.93 Å / % possible obs: 49.1 % / Rmerge(I) obs: 0.164 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.9→13 Å / Rfactor Rfree error: 0.006 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber Details: Electron density was weak for residues 111-116 and residues 220-226. In addition, residues 240-245 were not visible in the electron density and their coordinates are not included in this entry.
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 53.1361 Å2 / ksol: 0.458667 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.3 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→13 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→2.02 Å / Rfactor Rfree error: 0.017 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.9 / Classification: refinement | ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 13 Å / σ(F): 0 / % reflection Rfree: 7.2 % / Rfactor obs: 0.219 | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.286 / % reflection Rfree: 8 % / Rfactor Rwork: 0.224 |
