 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 3lgf | ||||||
|---|---|---|---|---|---|---|---|
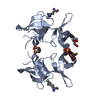
| タイトル | Crystal structure of the 53BP1 tandem tudor domain in complex with p53K370me2 | ||||||
 要素 要素 |
| ||||||
 キーワード キーワード |  CELL CYCLE (細胞周期) / tandem tudor domains / dimethylated p53 peptide / dna repair (DNA修復) / DNA damage (DNA修復) / DNA-binding / Methylation (メチル化) / Transcription (転写 (生物学)) / Transcription regulation CELL CYCLE (細胞周期) / tandem tudor domains / dimethylated p53 peptide / dna repair (DNA修復) / DNA damage (DNA修復) / DNA-binding / Methylation (メチル化) / Transcription (転写 (生物学)) / Transcription regulation | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報ubiquitin-modified histone reader activity / positive regulation of isotype switching / cellular response to X-ray / double-strand break repair via classical nonhomologous end joining / DNA repair complex / negative regulation of double-strand break repair via homologous recombination / telomeric DNA binding / SUMOylation of transcription factors / methylated histone binding / histone reader activity ...ubiquitin-modified histone reader activity / positive regulation of isotype switching / cellular response to X-ray / double-strand break repair via classical nonhomologous end joining / DNA repair complex / negative regulation of double-strand break repair via homologous recombination / telomeric DNA binding / SUMOylation of transcription factors / methylated histone binding / histone reader activity / DNA複製 / DNA damage checkpoint signaling / transcription coregulator activity / Nonhomologous End-Joining (NHEJ) / protein homooligomerization / G2/M DNA damage checkpoint / 動原体 / double-strand break repair via nonhomologous end joining / positive regulation of DNA-binding transcription factor activity / p53 binding / Recruitment and ATM-mediated phosphorylation of repair and signaling proteins at DNA double strand breaks / site of double-strand break / Processing of DNA double-strand break ends / histone binding / RNA polymerase II-specific DNA-binding transcription factor binding / damaged DNA binding / chromosome, telomeric region / nuclear body / DNA damage response / positive regulation of DNA-templated transcription / positive regulation of transcription by RNA polymerase II / 核質 / 細胞核 / 細胞質類似検索 - 分子機能 | ||||||
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) | ||||||
| 手法 | X線回折 / シンクロトロン / 分子置換 / 解像度: 1.5 Å | ||||||
 データ登録者 データ登録者 | Roy, S. / Kutateladze, T.G. | ||||||
 引用 引用 | ジャーナル: J.Mol.Biol. / 年: 2010 タイトル: Structural insight into p53 recognition by the 53BP1 tandem Tudor domain. 著者: Roy, S. / Musselman, C.A. / Kachirskaia, I. / Hayashi, R. / Glass, K.C. / Nix, J.C. / Gozani, O. / Appella, E. / Kutateladze, T.G. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 3lgf.cif.gz | 44.5 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb3lgf.ent.gz | 29.5 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 3lgf.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/lg/3lgfftp://data.pdbj.org/pub/pdb/validation_reports/lg/3lgf | HTTPS FTP |
|---|
-関連構造データ












-リンク
 PDBj
PDBj







- 集合体
集合体
| 登録構造単位 | 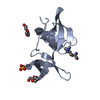
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| 2 |
| |||||||||||||||
| 単位格子 |
| |||||||||||||||
| Components on special symmetry positions |
|
-要素
| #1: タンパク質 | 分子量: 14029.840 Da / 分子数: 1 / 断片: Tandem tudor domains (RESIUDES 1484-1603) / 由来タイプ: 組換発現 / 由来: (組換発現) Homo sapiens (ヒト) / 遺伝子: TP53BP1 / プラスミド: pGEX6P1 / 発現宿主:  Escherichia coli (大腸菌) / 株 (発現宿主): BL21(DE3) / 参照: UniProt: Q12888 Escherichia coli (大腸菌) / 株 (発現宿主): BL21(DE3) / 参照: UniProt: Q12888 | ||||
|---|---|---|---|---|---|
| #2: タンパク質・ペプチド | 分子量: 1130.319 Da / 分子数: 1 / 由来タイプ: 合成 / 詳細: The peptide was chemically synthesized | ||||
| #3: 化合物 | ポリエチレングリコール  分子量: 150.173 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C6H14O4 分子量: 150.173 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C6H14O4#4: 化合物 | ChemComp-SO4 / | 硫酸塩  分子量: 96.063 Da / 分子数: 1 / 由来タイプ: 合成 / 式: SO4 分子量: 96.063 Da / 分子数: 1 / 由来タイプ: 合成 / 式: SO4#5: 水 | ChemComp-HOH / | 水 分子量: 18.015 Da / 分子数: 145 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 145 / 由来タイプ: 天然 / 式: H2O |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.24 Å3/Da / 溶媒含有率: 45.04 % |
|---|---|
| 結晶化 | 温度: 298 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 7 詳細: 0.1 M HEPES-Na pH 7.0, 2% PEG 400 and 2.4 M ammonium sulphate., VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-データ収集
| 回折 | 平均測定温度: 100 K |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: ALS  / ビームライン: 4.2.2 / 波長: 1 Å / ビームライン: 4.2.2 / 波長: 1 Å |
| 検出器 | タイプ: NOIR-1 / 検出器: CCD / 日付: 2009年1月10日 |
| 放射 | モノクロメーター: Double crystal / プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1 Å / 相対比: 1 |
| 反射 | 解像度: 1.5→39.05 Å / Num. all: 75227 / Num. obs: 21342 / % possible obs: 99.1 % / Observed criterion σ(F): 2 / 冗長度: 3.52 % / Rmerge(I) obs: 0.061 / Net I/σ(I): 9.1 |
| 反射 シェル | 解像度: 1.5→1.55 Å / 冗長度: 3.35 % / Rmerge(I) obs: 0.372 / Mean I/σ(I) obs: 2.5 / Num. unique all: 2136 / % possible all: 99.8 |
- 解析
解析
| ソフトウェア |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 分子置換 開始モデル: PDB entry 2G3R 解像度: 1.5→39.05 Å / SU ML: 0.51 / Isotropic thermal model: Isotropic / 交差検証法: THROUGHOUT / σ(F): 1.33 / 立体化学のターゲット値: Engh & Huber
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | 減衰半径: 0.9 Å / VDWプローブ半径: 1.11 Å / 溶媒モデル: FLAT BULK SOLVENT MODEL / Bsol: 56.114 Å2 / ksol: 0.36 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati sigma a obs: 0.208 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 1.5→39.05 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル |
|
