 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-22854 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Adeno-Associated Virus (AAV-DJ) - cryo-EM structure at 1.56 Angstrom Resolution | |||||||||
 マップデータ マップデータ | Full particle Relion map, sharpened using volume-whitening routine of cisTEM. Cropped version also provided for ease of comparison with coordinates. | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード |  Gene therapy vector / VIRUS LIKE PARTICLE (ウイルス様粒子) / VIRUS (ウイルス) Gene therapy vector / VIRUS LIKE PARTICLE (ウイルス様粒子) / VIRUS (ウイルス) | |||||||||
| 機能・相同性 | Phospholipase A2-like domain / Phospholipase A2-like domain / Parvovirus coat protein VP2 / Parvovirus coat protein VP1/VP2 / Parvovirus coat protein VP2 / Capsid/spike protein, ssDNA virus / T=1 icosahedral viral capsid / structural molecule activity / Capsid protein VP1 機能・相同性情報 機能・相同性情報 | |||||||||
| 生物種 |   Adeno-associated virus (アデノ随伴ウイルス) Adeno-associated virus (アデノ随伴ウイルス) | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 1.56 Å | |||||||||
 データ登録者 データ登録者 | Xie Q / Yoshioka CK | |||||||||
| 資金援助 |  米国, 1件 米国, 1件
| |||||||||
 引用 引用 | ジャーナル: Viruses / 年: 2020 タイトル: Adeno-Associated Virus (AAV-DJ)-Cryo-EM Structure at 1.56 Å Resolution. 著者: Qing Xie / Craig K Yoshioka / Michael S Chapman / 要旨: Adeno-associated virus is the leading viral vector for gene therapy. AAV-DJ is a recombinant variant developed for tropism to the liver. The AAV-DJ structure has been determined to 1.56 Å resolution ...Adeno-associated virus is the leading viral vector for gene therapy. AAV-DJ is a recombinant variant developed for tropism to the liver. The AAV-DJ structure has been determined to 1.56 Å resolution through cryo-electron microscopy (cryo-EM). Only apoferritin is reported in preprints at 1.6 Å or higher resolution, and AAV-DJ nearly matches the highest resolutions ever attained through X-ray diffraction of virus crystals. However, cryo-EM has the advantage that most of the hydrogens are clear, improving the accuracy of atomic refinement, and removing ambiguity in hydrogen bond identification. Outside of secondary structures where hydrogen bonding was predictable a priori, the networks of hydrogen bonds coming from direct observation of hydrogens and acceptor atoms are quite different from those inferred even at 2.8 Å resolution. The implications for understanding viral assembly mean that cryo-EM will likely become the favored approach for high resolution structural virology. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |
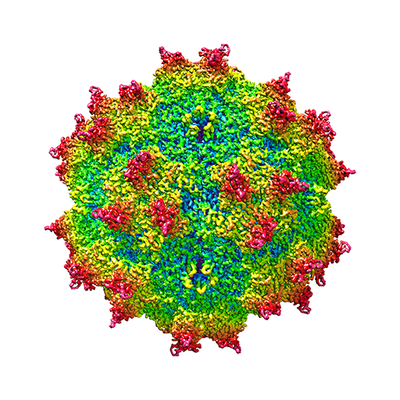
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_22854.map.gz | 765.5 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-22854-v30.xmlemd-22854.xml | 28.1 KB 28.1 KB | 表示 表示 | EMDBヘッダ |
| 画像 |  emd_22854.png emd_22854.png | 187.6 KB | ||
| マスクデータ | emd_22854_msk_1.map | 824 MB | マスクマップ | |
| Filedesc metadata | emd-22854.cif.gz | 7.7 KB | ||
| その他 | emd_22854_additional_1.map.gzemd_22854_additional_2.map.gzemd_22854_additional_3.map.gzemd_22854_half_map_1.map.gzemd_22854_half_map_2.map.gz | 752.3 MB 33.8 MB 34.7 MB 764.8 MB 764.7 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-22854ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22854 http://ftp.pdbj.org/pub/emdb/structures/EMD-22854ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22854 | HTTPS FTP |
-関連構造データ
| 関連構造データ |  7kfrMC M: このマップから作成された原子モデル C: 同じ文献を引用 ( |
|---|---|
| 類似構造データ | |
| 電子顕微鏡画像生データ | EMPIAR-10551 (タイトル: Adeno-Associated Virus (AAV-DJ)-Cryo-EM Structure at 1.56 Å Resolution Data size: 2.8 TB Data #1: Falcon3 frames converted to TIFF and compressed [micrographs - multiframe]) |










-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |



-マップ
| ファイル | ダウンロード / ファイル: emd_22854.map.gz / 形式: CCP4 / 大きさ: 824 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Full particle Relion map, sharpened using volume-whitening routine of cisTEM. Cropped version also provided for ease of comparison with coordinates. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 0.5105 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-添付データ
-マスク #1
| ファイル | emd_22854_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
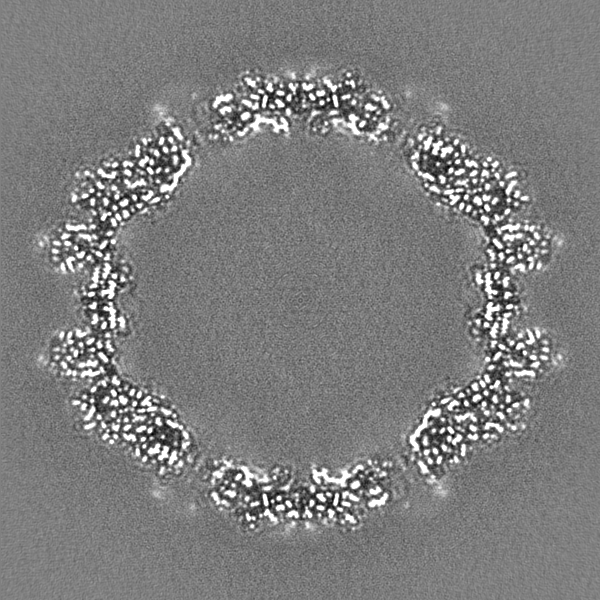
| 投影像・断面図 |
| ||||||||||||
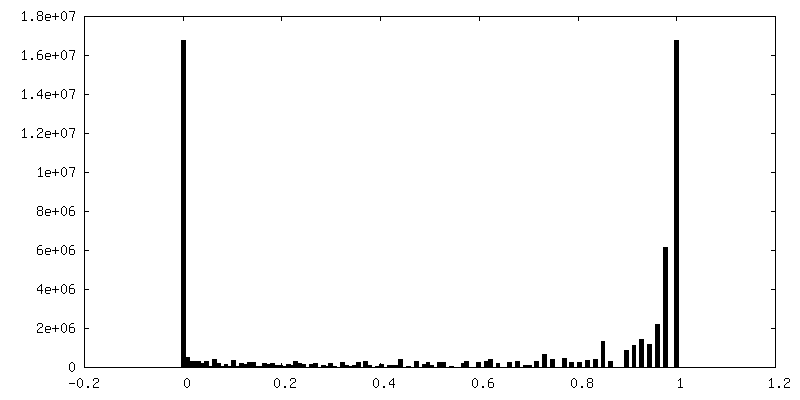
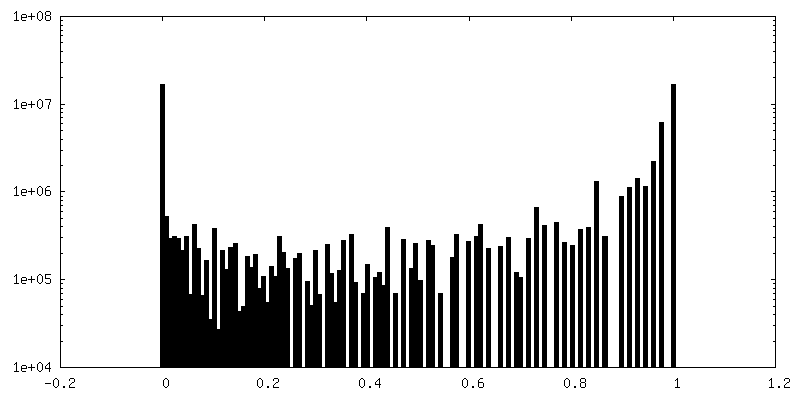
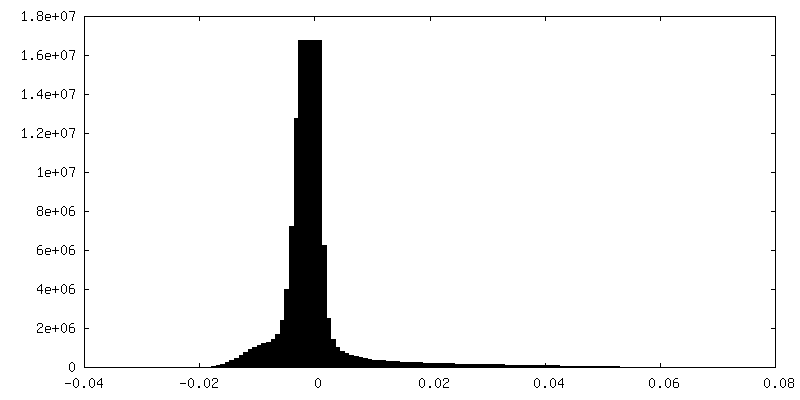
| 密度ヒストグラム |
 Z
Z Y
Y X
X







-追加マップ: Full particle, unsharpened Relion map, low pass filtered...
| ファイル | emd_22854_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Full particle, unsharpened Relion map, low pass filtered beyond FSC of 1.56 A. See also cropped version. | ||||||||||||
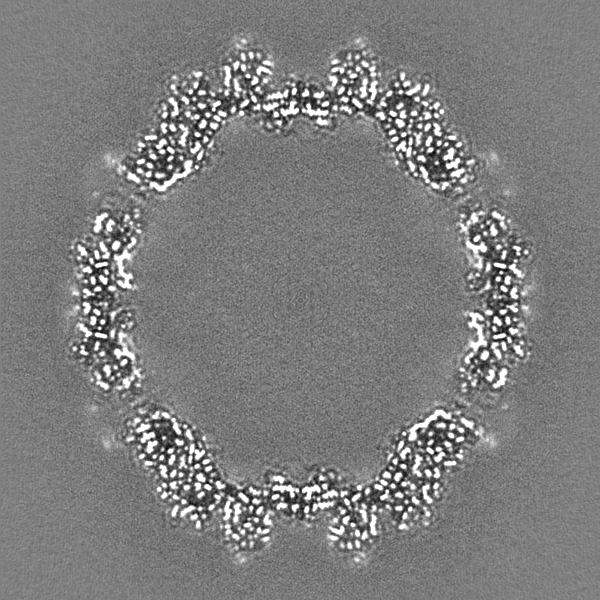
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |




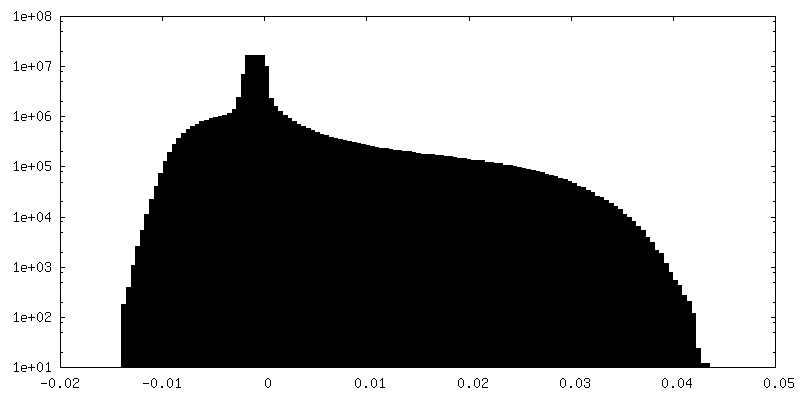
-追加マップ: Unsharpened Relion map, cropped ~20 Angstrom outside subunit...
| ファイル | emd_22854_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Unsharpened Relion map, cropped ~20 Angstrom outside subunit coordinates deposited, for ease of visualization. | ||||||||||||
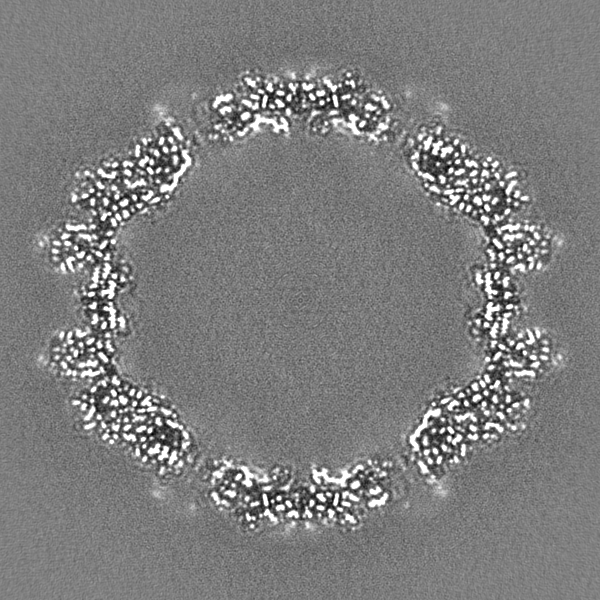
| 投影像・断面図 |
| ||||||||||||
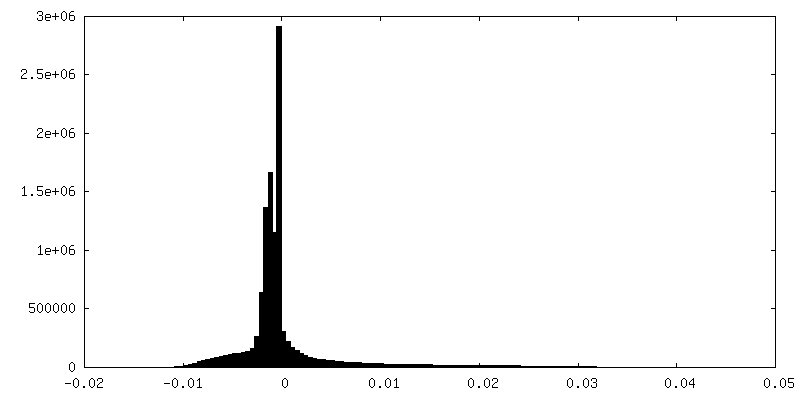
| 密度ヒストグラム |

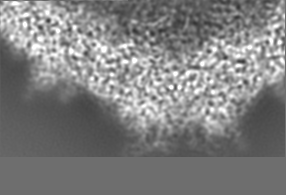
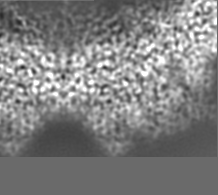
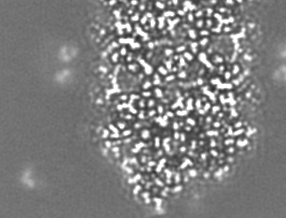
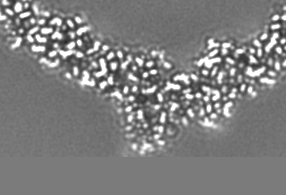
-追加マップ: Relion map, sharpened using volume-whitening routine of cisTEM,...
| ファイル | emd_22854_additional_3.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
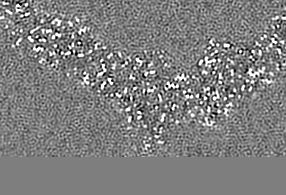
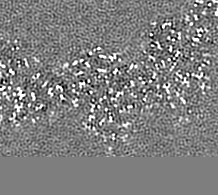
| 注釈 | Relion map, sharpened using volume-whitening routine of cisTEM, and cropped ~20 Angstrom outside deposited subunit coordinates for ease of comparison. | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
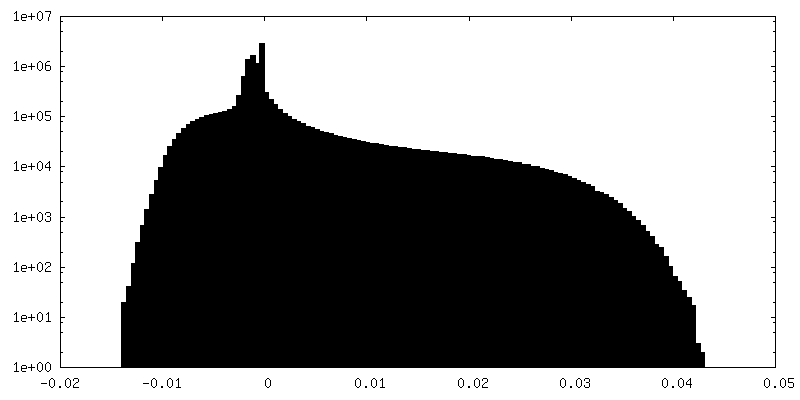
| 密度ヒストグラム |



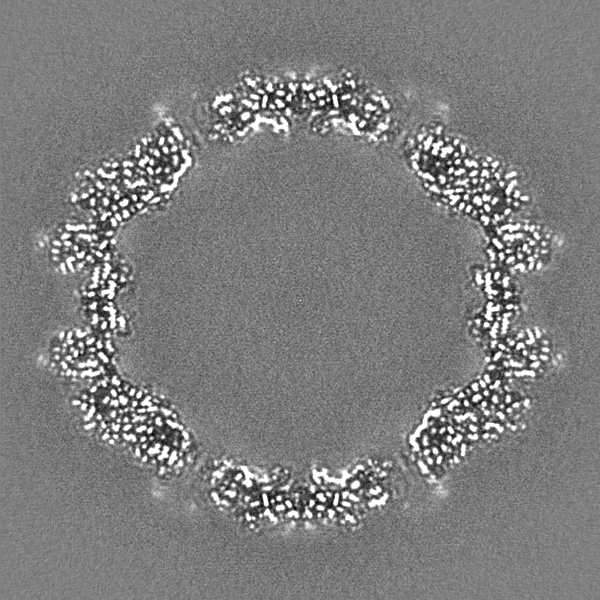
-ハーフマップ: Relion half map 1.
| ファイル | emd_22854_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Relion half map 1. | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |



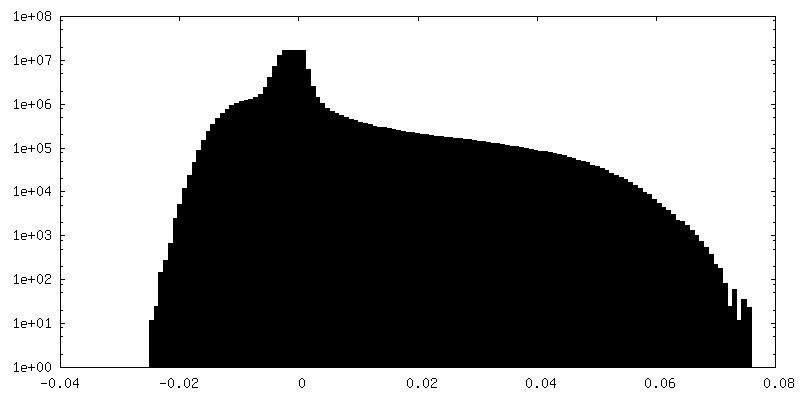
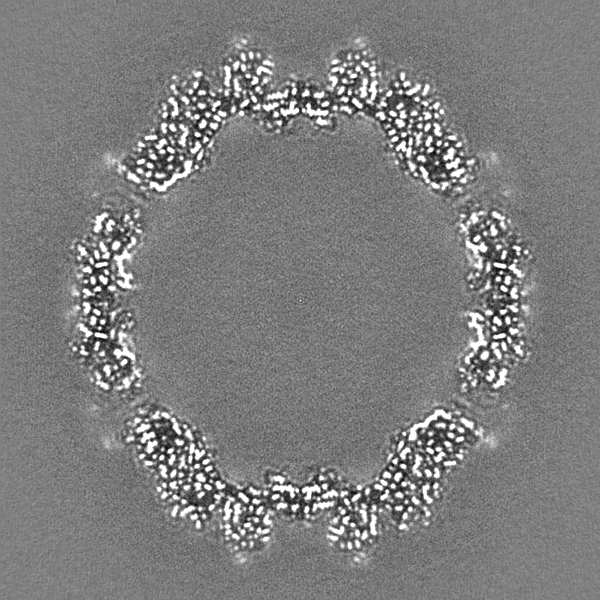
-ハーフマップ: Relion half map 2.
| ファイル | emd_22854_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Relion half map 2. | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |



- 試料の構成要素
試料の構成要素
-全体 : Adeno-associated virus
| 全体 | 名称: Adeno-associated virus (アデノ随伴ウイルス) |
|---|---|
| 要素 |
|
-超分子 #1: Adeno-associated virus
| 超分子 | 名称: Adeno-associated virus / タイプ: virus / ID: 1 / 親要素: 0 / 含まれる分子: #1 / NCBI-ID: 272636 / 生物種: Adeno-associated virus / ウイルスタイプ: VIRUS-LIKE PARTICLE / ウイルス・単離状態: STRAIN / ウイルス・エンベロープ: No / ウイルス・中空状態: Yes |
|---|---|
| 宿主 | 生物種:  Homo sapiens (ヒト) Homo sapiens (ヒト) |
| 分子量 | 理論値: 3.746 MDa |
| ウイルス殻 | Shell ID: 1 / 直径: 250.0 Å / T番号(三角分割数): 1 |
-分子 #1: Capsid protein VP1
| 分子 | 名称: Capsid protein VP1 / タイプ: protein_or_peptide / ID: 1 / コピー数: 1 / 光学異性体: LEVO |
|---|---|
| 由来(天然) | 生物種: Adeno-associated virus (アデノ随伴ウイルス) |
| 分子量 | 理論値: 58.877809 KDa |
| 組換発現 | 生物種:   Spodoptera frugiperda (ツマジロクサヨトウ) Spodoptera frugiperda (ツマジロクサヨトウ) |
| 配列 | 文字列: GADGVGNSSG NWHCDSTWMG DRVITTSTRT WALPTYNNHL YKQISNSTSG GSSNDNAYFG YSTPWGYFDF NRFHCHFSPR DWQRLINNN WGFRPKRLSF KLFNIQVKEV TQNEGTKTIA NNLTSTIQVF TDSEYQLPYV LGSAHQGCLP PFPADVFMIP Q YGYLTLNN ...文字列: GADGVGNSSG NWHCDSTWMG DRVITTSTRT WALPTYNNHL YKQISNSTSG GSSNDNAYFG YSTPWGYFDF NRFHCHFSPR DWQRLINNN WGFRPKRLSF KLFNIQVKEV TQNEGTKTIA NNLTSTIQVF TDSEYQLPYV LGSAHQGCLP PFPADVFMIP Q YGYLTLNN GSQAVGRSSF YCLEYFPSQM LRTGNNFQFT YTFEDVPFHS SYAHSQSLDR LMNPLIDQYL YYLSRTQTTG GT TNTQTLG FSQGGPNTMA NQAKNWLPGP CYRQQRVSKT SADNNNSEYS WTGATKYHLN GRDSLVNPGP AMASHKDDEE KFF PQSGVL IFGKQGSEKT NVDIEKVMIT DEEEIRTTNP VATEQYGSVS TNLQRGNRQA ATADVNTQGV LPGMVWQDRD VYLQ GPIWA KIPHTDGHFH PSPLMGGFGL KHPPPQILIK NTPVPADPPT TFNQSKLNSF ITQYSTGQVS VEIEWELQKE NSKRW NPEI QYTSNYYKST SVDFAVNTEG VYSEPRPIGT RYLTRNL UniProtKB: Capsid protein VP1 |
-分子 #2: MAGNESIUM ION
| 分子 | 名称: MAGNESIUM ION / タイプ: ligand / ID: 2 / コピー数: 3 / 式: MG |
|---|---|
| 分子量 | 理論値: 24.305 Da |
-分子 #3: water
| 分子 | 名称: water / タイプ: ligand / ID: 3 / コピー数: 265 / 式: HOH |
|---|---|
| 分子量 | 理論値: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 0.6 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 緩衝液 | pH: 7.4 構成要素:
| ||||||||||||
| グリッド | モデル: Quantifoil / 材質: COPPER / メッシュ: 200 / 前処理 - タイプ: GLOW DISCHARGE / 前処理 - 時間: 30 sec. / 前処理 - 雰囲気: OTHER / 前処理 - 気圧: 0.03 kPa 詳細: PELCO easiGlow Glow Discharge Cleaning System Current: 15mA | ||||||||||||
| 凍結 | 凍結剤: ETHANE / チャンバー内湿度: 100 % / チャンバー内温度: 293 K / 装置: FEI VITROBOT MARK IV 詳細: Two 3uL aliquots applied to grid (manual blotting between), prior to automated 3 second blot before plunging.. | ||||||||||||
| 詳細 | Monodisperse |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 電子線 | 加速電圧: 300 kV / 電子線源: FIELD EMISSION GUN |
| 電子光学系 | 倍率(補正後): 154400 / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / 最大 デフォーカス(公称値): -2.6 µm / 最小 デフォーカス(公称値): -0.8 µm / 倍率(公称値): 155000 |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER ホルダー冷却材: NITROGEN |
| 温度 | 最低: 93.0 K / 最高: 93.0 K |
| 詳細 | Coma-free alignment and objective astigmatism where corrected using Sherpa (Thermo Fisher, Inc.). |
| 撮影 | フィルム・検出器のモデル: FEI FALCON III (4k x 4k) 検出モード: INTEGRATING / デジタル化 - サイズ - 横: 4096 pixel / デジタル化 - サイズ - 縦: 4096 pixel / 撮影したグリッド数: 1 / 実像数: 2241 / 平均露光時間: 15.8 sec. / 平均電子線量: 30.0 e/Å2 詳細: Data were collected with pixel size of 0.514 angstrom on a FEI Titan Krios (Thermo Fisher, Inc.) at 300 kV, using a Falcon 3 camera (Thermo Fisher) with a total dose of approx. 30 e-/A2 ...詳細: Data were collected with pixel size of 0.514 angstrom on a FEI Titan Krios (Thermo Fisher, Inc.) at 300 kV, using a Falcon 3 camera (Thermo Fisher) with a total dose of approx. 30 e-/A2 fractionated across 200 frames. Camera dose rate was approx. 0.5 e-/pixel/s. Defocus was random in the nominal range of -0.8 to -2.6 um. Images were acquired in EPU (Thermo Fisher, Inc.) without using image shift. |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
-画像解析
| 粒子像選択 | 選択した数: 75316 詳細: DoG (Difference of Gaussian) Picker was used for initial automated particle selection. Templates were then generated by 2D classification, followed by particle template selection in Relion 3.0. |
|---|---|
| 初期モデル | モデルのタイプ: NONE |
| 初期 角度割当 | タイプ: MAXIMUM LIKELIHOOD / ソフトウェア - 名称: RELION (ver. 3.0) |
| 最終 3次元分類 | ソフトウェア - 名称: RELION (ver. 3.0) 詳細: Multiple rounds of 2D classification and 3D classification with C1 symmetry were used to remove outliers, resulting in 48,209 particles after deduplication. |
| 最終 角度割当 | タイプ: MAXIMUM LIKELIHOOD / ソフトウェア - 名称: RELION (ver. 3.0) |
| 最終 再構成 | 使用したクラス数: 1 / 想定した対称性 - 点群: I (正20面体型対称) / 解像度のタイプ: BY AUTHOR / 解像度: 1.56 Å / 解像度の算出法: FSC 0.143 CUT-OFF / ソフトウェア - 名称: RELION (ver. 3.0) 詳細: These 48209 particles refined to approx. 2.2 A with I1 symmetry, and subsequent refinements of beam tilt and per-particle CTF brought the resolution to 1.8 A. Further particle-polishing and ...詳細: These 48209 particles refined to approx. 2.2 A with I1 symmetry, and subsequent refinements of beam tilt and per-particle CTF brought the resolution to 1.8 A. Further particle-polishing and subsequent re-refinement of CTF brought the resolution to 1.70 A and a final reconstruction using Ewald sphere correction ended at 1.56 A. The map used for modeling was sharpened using the volume whitening routine in cisTEM. 使用した粒子像数: 48209 |
-原子モデル構築 1
| 初期モデル | PDB ID: Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| 詳細 | Stand-alone RSRef was used for refinement of magnification, resolution, envelope correction and atomic B-factors. This was alternated with RSRef-embedded CNS was used for molecular dynamics optimization (1st round) and stereochemically-restrained all-atom least-squares optimization. |
| 精密化 | 空間: REAL / プロトコル: FLEXIBLE FIT / 当てはまり具合の基準: Least-squares residual |
| 得られたモデル | PDB-7kfr: |

