National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
GM100888
United States
National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
GM140733
United States
National Institutes of Health/Office of the Director
OD017987
United States
National Institutes of Health/Office of the Director
OD023479
United States
National Institutes of Health/National Cancer Institute (NIH/NCI)
CA56036
United States
Citation
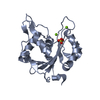
Journal: J Mol Biol / Year: 2022 Title: Terminase Subunits from the Pseudomonas-Phage E217. Authors: Ravi K Lokareddy / Chun-Feng David Hou / Steven G Doll / Fenglin Li / Richard E Gillilan / Francesca Forti / David S Horner / Federica Briani / Gino Cingolani / Abstract: Pseudomonas phages are increasingly important biomedicines for phage therapy, but little is known about how these viruses package DNA. This paper explores the terminase subunits from the Myoviridae ...Pseudomonas phages are increasingly important biomedicines for phage therapy, but little is known about how these viruses package DNA. This paper explores the terminase subunits from the Myoviridae E217, a Pseudomonas-phage used in an experimental cocktail to eradicate P. aeruginosa in vitro and in animal models. We identified the large (TerL) and small (TerS) terminase subunits in two genes ∼58 kbs away from each other in the E217 genome. TerL presents a classical two-domain architecture, consisting of an N-terminal ATPase and C-terminal nuclease domain arranged into a bean-shaped tertiary structure. A 2.05 Å crystal structure of the C-terminal domain revealed an RNase H-like fold with two magnesium ions in the nuclease active site. Mutations in TerL residues involved in magnesium coordination had a dominant-negative effect on phage growth. However, the two ions identified in the active site were too far from each other to promote two-metal-ion catalysis, suggesting a conformational change is required for nuclease activity. We also determined a 3.38 Å cryo-EM reconstruction of E217 TerS that revealed a ring-like decamer, departing from the most common nonameric quaternary structure observed thus far. E217 TerS contains both N-terminal helix-turn-helix motifs enriched in basic residues and a central channel lined with basic residues large enough to accommodate double-stranded DNA. Overexpression of TerS caused a more than a 4-fold reduction of E217 burst size, suggesting a catalytic amount of the protein is required for packaging. Together, these data expand the molecular repertoire of viral terminase subunits to Pseudomonas-phages used for phage therapy.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Pseudomonas phage vB_PaeM_E217 (virus)
Pseudomonas phage vB_PaeM_E217 (virus) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 5items
United States, 5items  Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads


 PDBj
PDBj Assembly
Assembly




 Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION
Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION Mass: 18.015 Da / Num. of mol.: 204 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 204 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing