 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7zrd: Cryo-EM map of the WT KdpFABC complex in the E1-P tight conformat... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7zrd | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM map of the WT KdpFABC complex in the E1-P tight conformation, stabilised with the inhibitor orthovanadate | ||||||||||||||||||
 Components Components | (Potassium-transporting ATPase ...) x 4 | ||||||||||||||||||
 Keywords Keywords | MEMBRANE PROTEIN / P-type ATPase / superfamily of K+ transporters (SKT) / potassium uptake system / off-cycle post-albers conformation | ||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationP-type K+ transporter / P-type potassium transmembrane transporter activity / potassium:proton antiporter complex / potassium ion-transporting ATPase complex / potassium ion binding / monoatomic cation transmembrane transport / potassium ion transmembrane transport / potassium ion transport / magnesium ion binding / ATP hydrolysis activity ...P-type K+ transporter / P-type potassium transmembrane transporter activity / potassium:proton antiporter complex / potassium ion-transporting ATPase complex / potassium ion binding / monoatomic cation transmembrane transport / potassium ion transmembrane transport / potassium ion transport / magnesium ion binding / ATP hydrolysis activity / ATP binding / plasma membrane Similarity search - Function | ||||||||||||||||||
| Biological species |  | ||||||||||||||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 3.3 Å | ||||||||||||||||||
 Authors Authors | Hielkema, L. / Stock, C. / Silberberg, J.M. / Corey, R.A. / Wunnicke, D. / Stansfeld, P.J. / Haenelt, I. / Paulino, C. | ||||||||||||||||||
| Funding support |  United Kingdom, 5items United Kingdom, 5items
| ||||||||||||||||||
 Citation Citation | Journal: Elife / Year: 2022 Title: Inhibited KdpFABC transitions into an E1 off-cycle state. Authors: Jakob M Silberberg / Charlott Stock / Lisa Hielkema / Robin A Corey / Jan Rheinberger / Dorith Wunnicke / Victor R A Dubach / Phillip J Stansfeld / Inga Hänelt / Cristina Paulino /   Abstract: KdpFABC is a high-affinity prokaryotic K uptake system that forms a functional chimera between a channel-like subunit (KdpA) and a P-type ATPase (KdpB). At high K levels, KdpFABC needs to be ...KdpFABC is a high-affinity prokaryotic K uptake system that forms a functional chimera between a channel-like subunit (KdpA) and a P-type ATPase (KdpB). At high K levels, KdpFABC needs to be inhibited to prevent excessive K accumulation to the point of toxicity. This is achieved by a phosphorylation of the serine residue in the TGES motif in the A domain of the pump subunit KdpB (KdpB). Here, we explore the structural basis of inhibition by KdpB phosphorylation by determining the conformational landscape of KdpFABC under inhibiting and non-inhibiting conditions. Under turnover conditions, we identified a new inhibited KdpFABC state that we termed E1P tight, which is not part of the canonical Post-Albers transport cycle of P-type ATPases. It likely represents the biochemically described stalled E1P state adopted by KdpFABC upon KdpB phosphorylation. The E1P tight state exhibits a compact fold of the three cytoplasmic domains and is likely adopted when the transition from high-energy E1P states to E2P states is unsuccessful. This study represents a structural characterization of a biologically relevant off-cycle state in the P-type ATPase family and supports the emerging discussion of P-type ATPase regulation by such states. | ||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7zrd.cif.gz | 267.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7zrd.ent.gz | 209.5 KB | Display | PDB format |
| PDBx/mmJSON format | 7zrd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zr/7zrdftp://data.pdbj.org/pub/pdb/validation_reports/zr/7zrd | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  14911MC  7zreC  7zrgC  7zrhC  7zriC  7zrjC  7zrkC  7zrlC  7zrmC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj
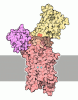


- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
-Potassium-transporting ATPase ... , 4 types, 4 molecules ACDB
| #1: Protein | Mass: 59218.613 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Protein | Mass: 20281.035 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
| #3: Protein/peptide | Mass: 3071.714 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
| #4: Protein | Mass: 72347.844 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
-Non-polymers , 3 types, 12 molecules 

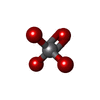
| #5: Chemical | ChemComp-K /  Mass: 39.098 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: K#6: Chemical |  Mass: 1464.043 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C81H156O17P2 / Comment: phospholipid*YM Mass: 1464.043 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C81H156O17P2 / Comment: phospholipid*YM#7: Chemical | ChemComp-VO4 / |  Mass: 114.939 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: VO4 / Feature type: SUBJECT OF INVESTIGATION Mass: 114.939 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: VO4 / Feature type: SUBJECT OF INVESTIGATION |
|---|
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: KdpFABC / Type: COMPLEX / Entity ID: #1-#4 / Source: RECOMBINANT |
|---|---|
| Molecular weight | Value: 0.157 MDa / Experimental value: NO |
| Source (natural) | Organism: |
| Source (recombinant) | Organism: |
| Buffer solution | pH: 8 Details: 10 mM Tris-HCl pH 8, 10 mM MgCl2, 10 mM NaCl and 0.0125% DDM |
| Specimen | Conc.: 4 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES |
| Specimen support | Details: at 5 mA / Grid material: GOLD / Grid mesh size: 300 divisions/in. / Grid type: Quantifoil R1.2/1.3 |
| Vitrification | Instrument: FEI VITROBOT MARK IV / Cryogen name: ETHANE-PROPANE / Humidity: 100 % / Chamber temperature: 293 K |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Talos Arctica / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: FEI TALOS ARCTICA |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD / Nominal magnification: 49407 X / Calibrated magnification: 49407 X / Nominal defocus max: 2000 nm / Nominal defocus min: 500 nm / Calibrated defocus min: 500 nm / Calibrated defocus max: 2000 nm / Cs: 2.7 mm / C2 aperture diameter: 100 µm / Alignment procedure: COMA FREE |
| Specimen holder | Cryogen: NITROGEN / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Temperature (max): 105 K / Temperature (min): 90 K |
| Image recording | Average exposure time: 9 sec. / Electron dose: 52 e/Å2 / Detector mode: COUNTING / Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Num. of grids imaged: 2 / Num. of real images: 2487 |
| EM imaging optics | Energyfilter name: GIF Bioquantum / Energyfilter slit width: 20 eV |
| Image scans | Width: 3838 / Height: 3710 / Movie frames/image: 60 / Used frames/image: 1-60 |
- Processing
Processing
| Software | Name: PHENIX / Version: 1.19.2_4158: / Classification: refinement | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EM software |
| ||||||||||||||||||||||||||||||||
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||||||||||
| Particle selection | Num. of particles selected: 164891 | ||||||||||||||||||||||||||||||||
| Symmetry | Point symmetry: C1 (asymmetric) | ||||||||||||||||||||||||||||||||
| 3D reconstruction | Resolution: 3.3 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 74927 / Algorithm: BACK PROJECTION / Symmetry type: POINT | ||||||||||||||||||||||||||||||||
| Atomic model building | Space: REAL | ||||||||||||||||||||||||||||||||
| Atomic model building | PDB-ID: 7NNM 7nnm Accession code: 7NNM / Source name: PDB / Type: experimental model | ||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
