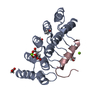
Journal: Nat Struct Mol Biol / Year: 2023 Title: Trapping the HIV-1 V3 loop in a helical conformation enables broad neutralization. Authors: Matthias Glögl / Nikolas Friedrich / Gabriele Cerutti / Thomas Lemmin / Young D Kwon / Jason Gorman / Liridona Maliqi / Peer R E Mittl / Maria C Hesselman / Daniel Schmidt / Jacqueline ...Authors: Matthias Glögl / Nikolas Friedrich / Gabriele Cerutti / Thomas Lemmin / Young D Kwon / Jason Gorman / Liridona Maliqi / Peer R E Mittl / Maria C Hesselman / Daniel Schmidt / Jacqueline Weber / Caio Foulkes / Adam S Dingens / Tatsiana Bylund / Adam S Olia / Raffaello Verardi / Thomas Reinberg / Nicolas S Baumann / Peter Rusert / Birgit Dreier / Lawrence Shapiro / Peter D Kwong / Andreas Plückthun / Alexandra Trkola / Abstract: The third variable (V3) loop on the human immunodeficiency virus 1 (HIV-1) envelope glycoprotein trimer is indispensable for virus cell entry. Conformational masking of V3 within the trimer allows ...The third variable (V3) loop on the human immunodeficiency virus 1 (HIV-1) envelope glycoprotein trimer is indispensable for virus cell entry. Conformational masking of V3 within the trimer allows efficient neutralization via V3 only by rare, broadly neutralizing glycan-dependent antibodies targeting the closed prefusion trimer but not by abundant antibodies that access the V3 crown on open trimers after CD4 attachment. Here, we report on a distinct category of V3-specific inhibitors based on designed ankyrin repeat protein (DARPin) technology that reinstitute the CD4-bound state as a key neutralization target with up to >90% breadth. Broadly neutralizing DARPins (bnDs) bound V3 solely on open envelope and recognized a four-turn amphipathic α-helix in the carboxy-terminal half of V3 (amino acids 314-324), which we termed 'αV3C'. The bnD contact surface on αV3C was as conserved as the CD4 binding site. Molecular dynamics and escape mutation analyses underscored the functional relevance of αV3C, highlighting the potential of αV3C-based inhibitors and, more generally, of postattachment inhibition of HIV-1.
Mass: 17160.244 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) synthetic construct (others) / Production host: Escherichia coli (E. coli)
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Human immunodeficiency virus 1
Human immunodeficiency virus 1 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Switzerland, 1items
Switzerland, 1items  Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads


 PDBj
PDBj






 Assembly
Assembly

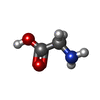
 Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H5NO2
Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H5NO2
 Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 18.015 Da / Num. of mol.: 153 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 153 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing