 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7z3w | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the AAL160 Fab | ||||||
 Components Components |
| ||||||
 Keywords Keywords | IMMUNE SYSTEM / monoclonal antibody | ||||||
| Function / homology | Immunoglobulins / Immunoglobulin-like / Sandwich / Mainly Beta / Chem-ETE Function and homology information Function and homology information | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2 Å | ||||||
 Authors Authors | Rondeau, J.M. / Lehmann, S. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: Commun Biol / Year: 2023 Title: "Redirecting an anti-IL-1 beta antibody to bind a new, unrelated and computationally predicted epitope on hIL-17A". Authors: Fischman, S. / Levin, I. / Rondeau, J.M. / Strajbl, M. / Lehmann, S. / Huber, T. / Nimrod, G. / Cebe, R. / Omer, D. / Kovarik, J. / Bernstein, S. / Sasson, Y. / Demishtein, A. / Shlamkovich, ...Authors: Fischman, S. / Levin, I. / Rondeau, J.M. / Strajbl, M. / Lehmann, S. / Huber, T. / Nimrod, G. / Cebe, R. / Omer, D. / Kovarik, J. / Bernstein, S. / Sasson, Y. / Demishtein, A. / Shlamkovich, T. / Bluvshtein, O. / Grossman, N. / Barak-Fuchs, R. / Zhenin, M. / Fastman, Y. / Twito, S. / Vana, T. / Zur, N. / Ofran, Y. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7z3w.cif.gz | 186.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7z3w.ent.gz | 147 KB | Display | PDB format |
| PDBx/mmJSON format | 7z3w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/z3/7z3wftp://data.pdbj.org/pub/pdb/validation_reports/z3/7z3w | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7z2mC  7z4tC  1mimS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24228.166 Da / Num. of mol.: 1 / Fragment: Fab heavy-chain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Cell (production host): hybridoma / Production host: Homo sapiens (human) | ||||||
|---|---|---|---|---|---|---|---|
| #2: Antibody | Mass: 23500.119 Da / Num. of mol.: 1 / Fragment: Fab light-chain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Cell (production host): Hybridoma / Production host: Homo sapiens (human) | ||||||
| #3: Chemical | ChemComp-ETE / 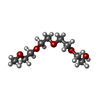  Mass: 208.252 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C9H20O5 Mass: 208.252 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C9H20O5#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 317 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 317 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | Has protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.62 Å3/Da / Density % sol: 66.02 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 9.5 / Details: 0.1M CHES pH 9.5, 50% (w/v) PEG 200 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM02 / Wavelength: 0.7998 Å / Beamline: BM02 / Wavelength: 0.7998 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MAR scanner 300 mm plate / Detector: IMAGE PLATE / Date: Jun 18, 1999 / Details: mirrors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.7998 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2→28 Å / Num. obs: 47440 / % possible obs: 99.9 % / Redundancy: 8.34 % / Biso Wilson estimate: 35.22 Å2 / Rmerge(I) obs: 0.05 / Χ2: 0.891 / Net I/σ(I): 11.7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1MIM.PDB Resolution: 2→16.38 Å / Cor.coef. Fo:Fc: 0.947 / Cor.coef. Fo:Fc free: 0.941 / SU R Cruickshank DPI: 0.131 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.138 / SU Rfree Blow DPI: 0.124 / SU Rfree Cruickshank DPI: 0.121
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 88.28 Å2 / Biso mean: 37.5 Å2 / Biso min: 23.73 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.25 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2→16.38 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.01 Å / Rfactor Rfree error: 0 / Total num. of bins used: 51
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
