 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 7s2h | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | Crystal structure of Trypanosoma cruzi glucokinase in the apo form (open conformation) | ||||||
 要素 要素 | Glucokinase 1, putative | ||||||
 キーワード キーワード | TRANSFERASE / Trypanosoma cruzi / Chagas disease | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報glucokinase / glucokinase activity / D-glucose binding / glycolytic process / ATP binding 類似検索 - 分子機能 | ||||||
| 生物種 |  | ||||||
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.8 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.8 Å | ||||||
 データ登録者 データ登録者 | Kearns, S.P. / Daneshian, L. / Swartz, P.D. / Carey, S.M. / Chruszcz, M. / D'Antonio, E.L. | ||||||
| 資金援助 | 1件
| ||||||
 引用 引用 | ジャーナル: To Be Published タイトル: Crystal structure of Trypanosoma cruzi glucokinase in the apo form (open conformation) 著者: Kearns, S.P. / Daneshian, L. / Swartz, P.D. / Carey, S.M. / Chruszcz, M. / D'Antonio, E.L. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 7s2h.cif.gz | 303.1 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb7s2h.ent.gz | 242.6 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 7s2h.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 7s2h_validation.pdf.gz | 462.2 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 7s2h_full_validation.pdf.gz | 464.5 KB | 表示 | |
| XML形式データ | 7s2h_validation.xml.gz | 32.6 KB | 表示 | |
| CIF形式データ | 7s2h_validation.cif.gz | 49.1 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/s2/7s2hftp://data.pdbj.org/pub/pdb/validation_reports/s2/7s2h | HTTPS FTP |
-関連構造データ
| 関連構造データ |  2q2rS S: 精密化の開始モデル |
|---|---|
| 類似構造データ |










-リンク
 PDBj
PDBj





- 集合体
集合体
| 登録構造単位 | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||
| 単位格子 |
| ||||||||||||||||||
| 非結晶学的対称性 (NCS) | NCSドメイン:
NCSドメイン領域: Component-ID: _ / Ens-ID: 1 / Beg auth comp-ID: MET / Beg label comp-ID: MET / End auth comp-ID: ASP / End label comp-ID: ASP / Refine code: _ / Auth seq-ID: -1 - 365 / Label seq-ID: 13 - 379
|
-要素
| #1: タンパク質 | 分子量: 42226.648 Da / 分子数: 2 / 由来タイプ: 組換発現 由来: (組換発現) 株: CL Brener / 遺伝子: Tc00.1047053510187.100 / 発現宿主:  #2: 化合物 | ChemComp-FLC / | 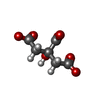  分子量: 189.100 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C6H5O7 分子量: 189.100 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C6H5O7#3: 化合物 | ChemComp-NA / |   分子量: 22.990 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Na 分子量: 22.990 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Na#4: 化合物 |   分子量: 96.063 Da / 分子数: 2 / 由来タイプ: 合成 / 式: SO4 分子量: 96.063 Da / 分子数: 2 / 由来タイプ: 合成 / 式: SO4#5: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 548 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 548 / 由来タイプ: 天然 / 式: H2O研究の焦点であるリガンドがあるか | N | |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.37 Å3/Da / 溶媒含有率: 48.1 % |
|---|---|
| 結晶化 | 温度: 295 K / 手法: 蒸気拡散法, シッティングドロップ法 / pH: 7.5 詳細: 1.0 uL of 7.4 mg/mL wt-TcGlcK in buffered solution [50 mM HEPES (pH 7.5), 0.2 M imidazole, 2 mM magnesium chloride] + 1.0 uL of precipitant solution [14% (w/v) PEG 3350, 0.1 M sodium citrate ...詳細: 1.0 uL of 7.4 mg/mL wt-TcGlcK in buffered solution [50 mM HEPES (pH 7.5), 0.2 M imidazole, 2 mM magnesium chloride] + 1.0 uL of precipitant solution [14% (w/v) PEG 3350, 0.1 M sodium citrate tribasic] was equilibrated against 85 uL of the precipitant solution using a 96-well sitting-drop plate (Innovadyne) |
-データ収集
| 回折 | 平均測定温度: 100 K / Serial crystal experiment: N |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: APS  / ビームライン: 22-ID / 波長: 1 Å / ビームライン: 22-ID / 波長: 1 Å |
| 検出器 | タイプ: RAYONIX MX300-HS / 検出器: CCD / 日付: 2014年6月27日 |
| 放射 | プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1 Å / 相対比: 1 |
| 反射 | 解像度: 1.8→40 Å / Num. obs: 72625 / % possible obs: 100 % / Observed criterion σ(I): -3 / 冗長度: 7.5 % / Rpim(I) all: 0.031 / Rrim(I) all: 0.083 / Net I/σ(I): 41.9 |
| 反射 シェル | 解像度: 1.8→1.83 Å / Mean I/σ(I) obs: 2.8 / Num. unique obs: 3637 / CC1/2: 0.833 / Rpim(I) all: 0.344 / Rrim(I) all: 0.949 |
- 解析
解析
| ソフトウェア |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 分子置換 開始モデル: 2Q2R 解像度: 1.8→35.92 Å / Cor.coef. Fo:Fc: 0.973 / Cor.coef. Fo:Fc free: 0.963 / SU B: 5.244 / SU ML: 0.077 / 交差検証法: THROUGHOUT / σ(F): 0 / ESU R: 0.114 / ESU R Free: 0.107 / 立体化学のターゲット値: MAXIMUM LIKELIHOOD 詳細: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : WITH TLS ADDED
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | イオンプローブ半径: 0.8 Å / 減衰半径: 0.8 Å / VDWプローブ半径: 1.2 Å / 溶媒モデル: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso max: 114.22 Å2 / Biso mean: 33.742 Å2 / Biso min: 20.92 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: final / 解像度: 1.8→35.92 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Ens-ID: 1 / 数: 11228 / Refine-ID: X-RAY DIFFRACTION / タイプ: interatomic distance / Rms dev position: 0.11 Å / Weight position: 0.05
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 1.8→1.847 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 TLS | 手法: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 TLSグループ |
|
