| 登録情報 | データベース: PDB / ID: 7q4r
|
|---|
| タイトル | Crystal structure of human HSP72-NBD in complex with fragment 1 |
|---|
 要素 要素 | Heat shock 70 kDa protein 1A |
|---|
 キーワード キーワード | CHAPERONE / Inhibitor / ATPase |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
: / denatured protein binding / cellular heat acclimation / negative regulation of inclusion body assembly / Viral RNP Complexes in the Host Cell Nucleus / death receptor agonist activity / C3HC4-type RING finger domain binding / positive regulation of nucleotide-binding oligomerization domain containing 2 signaling pathway / ATP-dependent protein disaggregase activity / positive regulation of microtubule nucleation ...: / denatured protein binding / cellular heat acclimation / negative regulation of inclusion body assembly / Viral RNP Complexes in the Host Cell Nucleus / death receptor agonist activity / C3HC4-type RING finger domain binding / positive regulation of nucleotide-binding oligomerization domain containing 2 signaling pathway / ATP-dependent protein disaggregase activity / positive regulation of microtubule nucleation / misfolded protein binding / negative regulation of mitochondrial outer membrane permeabilization involved in apoptotic signaling pathway / positive regulation of tumor necrosis factor-mediated signaling pathway / regulation of mitotic spindle assembly / aggresome / lysosomal transport / cellular response to steroid hormone stimulus / mRNA catabolic process / : / regulation of protein ubiquitination / Regulation of HSF1-mediated heat shock response / HSF1-dependent transactivation / cellular response to unfolded protein / response to unfolded protein / Mitochondrial unfolded protein response (UPRmt) / negative regulation of extrinsic apoptotic signaling pathway in absence of ligand / Attenuation phase / chaperone-mediated protein complex assembly / negative regulation of endoplasmic reticulum stress-induced intrinsic apoptotic signaling pathway / ATP metabolic process / transcription regulator inhibitor activity / inclusion body / heat shock protein binding / negative regulation of protein ubiquitination / centriole / protein folding chaperone / HSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand / positive regulation of erythrocyte differentiation / positive regulation of RNA splicing / positive regulation of interleukin-8 production / AUF1 (hnRNP D0) binds and destabilizes mRNA / negative regulation of transforming growth factor beta receptor signaling pathway / ATP-dependent protein folding chaperone / G protein-coupled receptor binding / negative regulation of cell growth / positive regulation of NF-kappaB transcription factor activity / PKR-mediated signaling / histone deacetylase binding / disordered domain specific binding / transcription corepressor activity / unfolded protein binding / positive regulation of proteasomal ubiquitin-dependent protein catabolic process / cellular response to heat / virus receptor activity / protein refolding / cellular response to oxidative stress / blood microparticle / vesicle / ficolin-1-rich granule lumen / protein stabilization / nuclear speck / cadherin binding / receptor ligand activity / ribonucleoprotein complex / signaling receptor binding / negative regulation of cell population proliferation / focal adhesion / centrosome / ubiquitin protein ligase binding / Neutrophil degranulation / positive regulation of gene expression / negative regulation of apoptotic process / perinuclear region of cytoplasm / enzyme binding / endoplasmic reticulum / negative regulation of transcription by RNA polymerase II / protein-containing complex / ATP hydrolysis activity / mitochondrion / extracellular space / RNA binding / extracellular exosome / extracellular region / nucleoplasm / ATP binding / nucleus / plasma membrane / cytosol / cytoplasm類似検索 - 分子機能 Heat shock hsp70 proteins family signature 2. / Heat shock hsp70 proteins family signature 1. / Heat shock hsp70 proteins family signature 3. / Heat shock protein 70, conserved site / Heat shock protein 70kD, peptide-binding domain superfamily / Heat shock protein 70 family / Hsp70 protein / Heat shock protein 70kD, C-terminal domain superfamily / ATPase, substrate binding domain, subdomain 4 / Actin; Chain A, domain 4 ...Heat shock hsp70 proteins family signature 2. / Heat shock hsp70 proteins family signature 1. / Heat shock hsp70 proteins family signature 3. / Heat shock protein 70, conserved site / Heat shock protein 70kD, peptide-binding domain superfamily / Heat shock protein 70 family / Hsp70 protein / Heat shock protein 70kD, C-terminal domain superfamily / ATPase, substrate binding domain, subdomain 4 / Actin; Chain A, domain 4 / ATPase, nucleotide binding domain / Alpha-Beta Complex / Alpha Beta類似検索 - ドメイン・相同性 Chem-8X6 / PHOSPHATE ION / Heat shock 70 kDa protein 1A類似検索 - 構成要素 |
|---|
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.79 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.79 Å |
|---|
 データ登録者 データ登録者 | Le Bihan, Y.V. / Westwood, I.M. / van Montfort, R.L.M. |
|---|
| 資金援助 |  英国, 1件 英国, 1件 | 組織 | 認可番号 | 国 |
|---|
| Cancer Research UK | C309/A11566 | 英国 |
|
|---|
 引用 引用 | ジャーナル: Molecules / 年: 2022
タイトル: Discovery and Characterization of a Cryptic Secondary Binding Site in the Molecular Chaperone HSP70.
著者: O'Connor, S. / Le Bihan, Y.V. / Westwood, I.M. / Liu, M. / Mak, O.W. / Zazeri, G. / Povinelli, A.P.R. / Jones, A.M. / van Montfort, R. / Reynisson, J. / Collins, I. |
|---|
| 履歴 | | 登録 | 2021年11月2日 | 登録サイト: PDBE / 処理サイト: PDBE |
|---|
| 改定 1.0 | 2022年2月2日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2022年3月2日 | Group: Database references / カテゴリ: citation / Item: _citation.pdbx_database_id_PubMed / _citation.title |
|---|
| 改定 1.2 | 2024年1月31日 | Group: Data collection / Refinement description
カテゴリ: chem_comp_atom / chem_comp_bond / pdbx_initial_refinement_model |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード










 PDBj
PDBj








 集合体
集合体


 分子量: 24.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Mg
分子量: 24.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Mg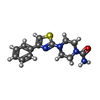
 分子量: 288.368 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C14H16N4OS / タイプ: SUBJECT OF INVESTIGATION
分子量: 288.368 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C14H16N4OS / タイプ: SUBJECT OF INVESTIGATION
 分子量: 94.971 Da / 分子数: 1 / 由来タイプ: 合成 / 式: PO4
分子量: 94.971 Da / 分子数: 1 / 由来タイプ: 合成 / 式: PO4 分子量: 18.015 Da / 分子数: 409 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 409 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析