 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7p9t | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of CD73 in complex with dCMP in the open form | ||||||
 Components Components | 5'-nucleotidase | ||||||
 Keywords Keywords | HYDROLASE / ecto-5'-nucleotidase / eN / substrate | ||||||
| Function / homology |  Function and homology information Function and homology informationthymidylate 5'-phosphatase / ADP catabolic process / 5'-deoxynucleotidase / 5'-deoxynucleotidase activity / 7-methylguanosine nucleotidase / adenosine biosynthetic process / inhibition of non-skeletal tissue mineralization / Pyrimidine catabolism / IMP-specific 5'-nucleotidase / AMP catabolic process ...thymidylate 5'-phosphatase / ADP catabolic process / 5'-deoxynucleotidase / 5'-deoxynucleotidase activity / 7-methylguanosine nucleotidase / adenosine biosynthetic process / inhibition of non-skeletal tissue mineralization / Pyrimidine catabolism / IMP-specific 5'-nucleotidase / AMP catabolic process / Purine catabolism / 5'-nucleotidase / 5'-nucleotidase activity / Nicotinate metabolism / leukocyte cell-cell adhesion / DNA metabolic process / response to ATP / calcium ion homeostasis / ATP metabolic process / Purinergic signaling in leishmaniasis infection / negative regulation of inflammatory response / external side of plasma membrane / nucleotide binding / cell surface / extracellular exosome / nucleoplasm / zinc ion binding / membrane / identical protein binding / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.79 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.79 Å | ||||||
 Authors Authors | Scaletti, E.R. / Strater, N. | ||||||
 Citation Citation | Journal: Purinergic Signal / Year: 2021 Title: Substrate binding modes of purine and pyrimidine nucleotides to human ecto-5'-nucleotidase (CD73) and inhibition by their bisphosphonic acid derivatives. Authors: Scaletti, E. / Huschmann, F.U. / Mueller, U. / Weiss, M.S. / Strater, N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7p9t.cif.gz | 227 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7p9t.ent.gz | 178.9 KB | Display | PDB format |
| PDBx/mmJSON format | 7p9t.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p9/7p9tftp://data.pdbj.org/pub/pdb/validation_reports/p9/7p9t | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7p9nC  7p9rC  7pa4C  7pb5C  7pbaC  7pbbC  7pbyC  7pcpC  7pd9C  4h2gS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 60464.340 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: NT5E, NT5, NTE / Production host:  | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#3: Chemical | ChemComp-CA / |   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca#4: Chemical | ChemComp-DCM / | 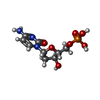  Mass: 307.197 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N3O7P / Feature type: SUBJECT OF INVESTIGATION Mass: 307.197 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N3O7P / Feature type: SUBJECT OF INVESTIGATION#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 226 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 226 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.52 Å3/Da / Density % sol: 51.22 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, hanging drop Details: 7 mg/mL protein concentration, 100 mM Tris pH 7.8, 10 % PEG6000, equal amounts of protein and reservoir. Following crystal formation (1-2 days), the crystals were transferred to soaking ...Details: 7 mg/mL protein concentration, 100 mM Tris pH 7.8, 10 % PEG6000, equal amounts of protein and reservoir. Following crystal formation (1-2 days), the crystals were transferred to soaking solution containing reservoir solution and 50 mM dCMP. Crystals were then transferred to cryo solution containing an additional 20 % glycerol, soaked for ~2-5 min, and flash frozen in liquid nitrogen. |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.8943 Å / Beamline: 14.1 / Wavelength: 0.8943 Å | ||||||||||||||||||||||||
| Detector | Type: RAYONIX MX-225 / Detector: CCD / Date: Oct 4, 2016 | ||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.8943 Å / Relative weight: 1 | ||||||||||||||||||||||||
| Reflection | Resolution: 1.79→47.95 Å / Num. obs: 58377 / % possible obs: 99.7 % / Redundancy: 4.1 % / CC1/2: 0.997 / Rmerge(I) obs: 0.107 / Rpim(I) all: 0.059 / Rrim(I) all: 0.122 / Net I/σ(I): 11.1 | ||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: 4h2g Resolution: 1.79→47.95 Å / Cor.coef. Fo:Fc: 0.954 / Cor.coef. Fo:Fc free: 0.935 / SU B: 6.14 / SU ML: 0.083 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.116 / ESU R Free: 0.112 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT U VALUES : WITH TLS ADDED
| |||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 67.68 Å2 / Biso mean: 19.897 Å2 / Biso min: 9.4 Å2
| |||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.79→47.95 Å
| |||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.79→1.836 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 25.11 Å / Origin y: 24.935 Å / Origin z: 23.366 Å
|
