 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7nre: Crystal structure of E.coli BamA beta-barrel in complex with daro... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7nre | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of E.coli BamA beta-barrel in complex with darobactin (crystal form 1) | ||||||
 Components Components |
| ||||||
 Keywords Keywords | MEMBRANE PROTEIN / Beta-Barrel / outer membrane / protein insertion / protein folding / protein maturation / antibiotic / natural product / cyclized peptide | ||||||
| Function / homology |  Function and homology information Function and homology informationBam protein complex / Gram-negative-bacterium-type cell outer membrane assembly / protein insertion into membrane Similarity search - Function | ||||||
| Biological species |  synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Jakob, R.P. / Kaur, H. / Marzinek, J.K. / Green, R. / Imai, Y. / Bolla, J. / Robinson, C. / Bond, P.J. / Lewis, K. / Maier, T. / Hiller, S. | ||||||
 Citation Citation | Journal: Nature / Year: 2021 Title: The antibiotic darobactin mimics a β-strand to inhibit outer membrane insertase. Authors: Hundeep Kaur / Roman P Jakob / Jan K Marzinek / Robert Green / Yu Imai / Jani Reddy Bolla / Elia Agustoni / Carol V Robinson / Peter J Bond / Kim Lewis / Timm Maier / Sebastian Hiller /     Abstract: Antibiotics that target Gram-negative bacteria in new ways are needed to resolve the antimicrobial resistance crisis. Gram-negative bacteria are protected by an additional outer membrane, rendering ...Antibiotics that target Gram-negative bacteria in new ways are needed to resolve the antimicrobial resistance crisis. Gram-negative bacteria are protected by an additional outer membrane, rendering proteins on the cell surface attractive drug targets. The natural compound darobactin targets the bacterial insertase BamA-the central unit of the essential BAM complex, which facilitates the folding and insertion of outer membrane proteins. BamA lacks a typical catalytic centre, and it is not obvious how a small molecule such as darobactin might inhibit its function. Here we resolve the mode of action of darobactin at the atomic level using a combination of cryo-electron microscopy, X-ray crystallography, native mass spectrometry, in vivo experiments and molecular dynamics simulations. Two cyclizations pre-organize the darobactin peptide in a rigid β-strand conformation. This creates a mimic of the recognition signal of native substrates with a superior ability to bind to the lateral gate of BamA. Upon binding, darobactin replaces a lipid molecule from the lateral gate to use the membrane environment as an extended binding pocket. Because the interaction between darobactin and BamA is largely mediated by backbone contacts, it is particularly robust against potential resistance mutations. Our results identify the lateral gate as a functional hotspot in BamA and will allow the rational design of antibiotics that target this bacterial Achilles heel. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7nre.cif.gz | 230.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7nre.ent.gz | 186.7 KB | Display | PDB format |
| PDBx/mmJSON format | 7nre.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nr/7nreftp://data.pdbj.org/pub/pdb/validation_reports/nr/7nre | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7nrfC  7nriC  6qgwS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 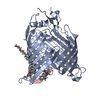
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 46022.789 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Protein/peptide | 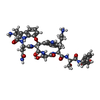  Type: Peptide-like / Class: Antibiotic / Mass: 971.047 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) / References: Darobactin Type: Peptide-like / Class: Antibiotic / Mass: 971.047 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) / References: Darobactin | ||||||||
| #3: Chemical | ChemComp-C8E / (   Mass: 306.438 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C16H34O5 / Comment: C8E, detergent*YM Mass: 306.438 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C16H34O5 / Comment: C8E, detergent*YM#4: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 201 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 201 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Nonpolymer details | Original Refinement was done with Darobactin defined as one ligand. | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.75 Å3/Da / Density % sol: 55.23 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: 0.06 M Magnesium chloride/Calcium Chloride, 0.1 M imidazole/2-(N-morpholino)ethanesulfonic acid pH 6.5, 12.5% v/v 2-Methyl-2,4-pentanediol, 12.5% w/v Polyethylene glycol 1,000, and 12.5% w/v ...Details: 0.06 M Magnesium chloride/Calcium Chloride, 0.1 M imidazole/2-(N-morpholino)ethanesulfonic acid pH 6.5, 12.5% v/v 2-Methyl-2,4-pentanediol, 12.5% w/v Polyethylene glycol 1,000, and 12.5% w/v Polyethylene glycol 3,350 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS / Beamline: X06SA / Wavelength: 1.000009 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Jul 19, 2019 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.000009 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→46.602 Å / Num. obs: 22630 / % possible obs: 99.7 % / Redundancy: 11.7 % / CC1/2: 0.997 / Rmerge(I) obs: 0.156 / Rpim(I) all: 0.072 / Rrim(I) all: 0.172 / Net I/σ(I): 7.1 |
| Reflection shell | Resolution: 2.3→2.38 Å / Redundancy: 8.7 % / Rmerge(I) obs: 1.456 / Mean I/σ(I) obs: 1.3 / Num. unique obs: 2230 / CC1/2: 0.535 / Rpim(I) all: 0.66 / Rrim(I) all: 1.602 / % possible all: 98.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6QGW Resolution: 2.3→46.602 Å / SU ML: 0.33 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 25.92
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 158.33 Å2 / Biso mean: 49.6919 Å2 / Biso min: 19.08 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.3→46.602 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
