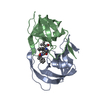
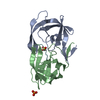
登録情報 データベース : PDB / ID : 7n6vタイトル Crystal structure of HIV-1 Protease multiple mutants PRS17 with Revertant mutation V48G bound to inhibitor Amprenavir Protease キーワード / / / 機能・相同性 分子機能 ドメイン・相同性 構成要素
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / 生物種 手法 / / / 解像度 : 1.39 Å データ登録者 Burnaman, S.H. / Wang, Y.-F. / Weber, I.T. 資金援助 組織 認可番号 国 National Institutes of Health/National Institute Of Allergy and Infectious Diseases (NIH/NIAID) AI150461
ジャーナル : J.Mol.Graph.Model. / 年 : 2021タイトル : Revertant mutation V48G alters conformational dynamics of highly drug resistant HIV protease PRS17.著者 : Burnaman, S.H. / Kneller, D.W. / Wang, Y.F. / Kovalevsky, A. / Weber, I.T. 履歴 登録 2021年6月9日 登録サイト / 処理サイト 改定 1.0 2021年9月8日 Provider / タイプ 改定 1.1 2023年10月18日 Group / Refinement descriptionカテゴリ / chem_comp_bond / pdbx_initial_refinement_model
すべて表示 表示を減らす
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Human immunodeficiency virus type 1 group M subtype B (ヒト免疫不全ウイルス)
Human immunodeficiency virus type 1 group M subtype B (ヒト免疫不全ウイルス) X線回折 /
X線回折 /  データ登録者
データ登録者 米国, 1件
米国, 1件  引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード

























 PDBj
PDBj


 集合体
集合体

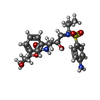
 分子量: 505.627 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C25H35N3O6S / タイプ: SUBJECT OF INVESTIGATION / コメント: プロテアーゼ阻害剤, 薬剤*YM
分子量: 505.627 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C25H35N3O6S / タイプ: SUBJECT OF INVESTIGATION / コメント: プロテアーゼ阻害剤, 薬剤*YM
 分子量: 92.094 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C3H8O3
分子量: 92.094 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C3H8O3 分子量: 18.015 Da / 分子数: 85 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 85 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析