 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7l4a: Crystal Structure of Cytidylate kinase from Encephalitozoon cunic... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7l4a | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Cytidylate kinase from Encephalitozoon cuniculi GB-M1 in complex with two CDP molecules | ||||||
 Components Components | Cytidylate kinase | ||||||
 Keywords Keywords | TRANSFERASE / SSGCID / cytidylate kinase / Encephalitozoon cuniculi / CDP / CTP / Structural Genomics / Seattle Structural Genomics Center for Infectious Disease | ||||||
| Function / homology |  Function and homology information Function and homology information(d)CMP kinase / CMP kinase activity / dCMP kinase activity / nucleobase-containing small molecule interconversion / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |  Encephalitozoon cuniculi (fungus) Encephalitozoon cuniculi (fungus) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 1.5 Å | ||||||
 Authors Authors | Seattle Structural Genomics Center for Infectious Disease (SSGCID) | ||||||
 Citation Citation | Journal: to be published Title: Crystal Structure of Cytidylate kinase from Encephalitozoon cuniculi GB-M1 in complex with two CDP molecules Authors: Abendroth, J.A. / Fox III, D. / Lorimer, D.D. / Horanyi, P.S. / Edwards, T.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7l4a.cif.gz | 138.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7l4a.ent.gz | 89.1 KB | Display | PDB format |
| PDBx/mmJSON format | 7l4a.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 7l4a_validation.pdf.gz | 1.4 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 7l4a_full_validation.pdf.gz | 1.4 MB | Display | |
| Data in XML | 7l4a_validation.xml.gz | 13.1 KB | Display | |
| Data in CIF | 7l4a_validation.cif.gz | 19.4 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/l4/7l4aftp://data.pdbj.org/pub/pdb/validation_reports/l4/7l4a | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|




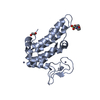





-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 26337.838 Da / Num. of mol.: 1 / Fragment: EncuA.01086.a.AE1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Encephalitozoon cuniculi (strain GB-M1) (fungus)Strain: GB-M1 / Gene: ECU03_1270 / Plasmid: Endua.01086.a.aE1 / Production host:  | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | 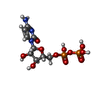  Mass: 403.176 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H15N3O11P2 / Feature type: SUBJECT OF INVESTIGATION Mass: 403.176 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H15N3O11P2 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | ChemComp-SO4 / |   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-EDO / |   Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 235 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 235 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.15 Å3/Da / Density % sol: 42.8 % |
|---|---|
| Crystal grow | Temperature: 287 K / Method: vapor diffusion, sitting drop / pH: 5.5 Details: Optimization condition: 100mM BisTris pH 6, 200mM ammonium sulfate, 26% (w/V) PEG 3350: EucuA.01086.a.AE1.PS38633 at 56.56mg/ml + 5mM CTP + 5mM MgCl2: tray 318933 a2: cryo: 20% EG + ligands: ...Details: Optimization condition: 100mM BisTris pH 6, 200mM ammonium sulfate, 26% (w/V) PEG 3350: EucuA.01086.a.AE1.PS38633 at 56.56mg/ml + 5mM CTP + 5mM MgCl2: tray 318933 a2: cryo: 20% EG + ligands: puck akp1-5. For phasing, a crystal from MCSG1, condition D7 (20% (w/V) PEG 3000, 100mM sodium citrate tribasic / citric acid pH 5.5: EncuA.01086.a.AE1.PS38636 at 28.28mg/ml, tray 315976 d7) was dipped for 20sec in a solution of 4ul half saturated NaI in ethylene glycol and reservoir and directly vitrified. This crystal form could not be reproduced. |
-Data collection
| Diffraction |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.5→50 Å / Num. obs: 35784 / % possible obs: 99.5 % / Redundancy: 4.026 % / Biso Wilson estimate: 25.36 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.042 / Rrim(I) all: 0.049 / Χ2: 0.948 / Net I/σ(I): 17.71 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|

- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 1.5→44.8 Å / SU ML: 0.1657 / Cross valid method: FREE R-VALUE / σ(F): 1.36 / Phase error: 20.4856 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 25.13 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→44.8 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
