 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7kcb: Symmetry in Yeast Alcohol Dehydrogenase 1 -Closed Form with NAD+ ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7kcb | ||||||
|---|---|---|---|---|---|---|---|
| Title | Symmetry in Yeast Alcohol Dehydrogenase 1 -Closed Form with NAD+ and Trifluoroethanol | ||||||
 Components Components | ADH1 isoform 1 | ||||||
 Keywords Keywords | OXIDOREDUCTASE / Alcohol dehydrogenase / holo-enzyme complex | ||||||
| Function / homology | TRIFLUOROETHANOL / NICOTINAMIDE-ADENINE-DINUCLEOTIDE / :  Function and homology information Function and homology information | ||||||
| Biological species |  | ||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 2.77 Å | ||||||
 Authors Authors | Subramanian, R. / Chang, L. / Li, Z. / Plapp, B.V. / Guntupalli, S.R. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2021 Title: Cryo-Electron Microscopy Structures of Yeast Alcohol Dehydrogenase. Authors: Sai Rohit Guntupalli / Zhuang Li / Leifu Chang / Bryce V Plapp / Ramaswamy Subramanian /   Abstract: Structures of yeast alcohol dehydrogenase determined by X-ray crystallography show that the subunits have two different conformational states in each of the two dimers that form the tetramer. ...Structures of yeast alcohol dehydrogenase determined by X-ray crystallography show that the subunits have two different conformational states in each of the two dimers that form the tetramer. Apoenzyme and holoenzyme complexes relevant to the catalytic mechanism were described, but the asymmetry led to questions about the cooperativity of the subunits in catalysis. This study used cryo-electron microscopy (cryo-EM) to provide structures for the apoenzyme, two different binary complexes with NADH, and a ternary complex with NAD and 2,2,2-trifluoroethanol. All four subunits in each of these complexes are identical, as the tetramers have 2 symmetry, suggesting that there is no preexisting asymmetry and that the subunits can be independently active. The apoenzyme and one enzyme-NADH complex have "open" conformations and the inverted coordination of the catalytic zinc with Cys-43, His-66, Glu-67, and Cys-153, whereas another enzyme-NADH complex and the ternary complex have closed conformations with the classical coordination of the zinc with Cys-43, His-66, Cys-153, and a water or the oxygen of trifluoroethanol. The conformational change involves interactions of Arg-340 with the pyrophosphate group of the coenzyme and Glu-67. The cryo-EM and X-ray crystallography studies provide structures relevant for the catalytic mechanism. #1: Journal: Biochemistry / Year: 2014Title: Yeast alcohol dehydrogenase structure and catalysis. Authors: Savarimuthu Baskar Raj / S Ramaswamy / Bryce V Plapp / Abstract: Yeast (Saccharomyces cerevisiae) alcohol dehydrogenase I (ADH1) is the constitutive enzyme that reduces acetaldehyde to ethanol during the fermentation of glucose. ADH1 is a homotetramer of subunits ...Yeast (Saccharomyces cerevisiae) alcohol dehydrogenase I (ADH1) is the constitutive enzyme that reduces acetaldehyde to ethanol during the fermentation of glucose. ADH1 is a homotetramer of subunits with 347 amino acid residues. A structure for ADH1 was determined by X-ray crystallography at 2.4 Å resolution. The asymmetric unit contains four different subunits, arranged as similar dimers named AB and CD. The unit cell contains two different tetramers made up of "back-to-back" dimers, AB:AB and CD:CD. The A and C subunits in each dimer are structurally similar, with a closed conformation, bound coenzyme, and the oxygen of 2,2,2-trifluoroethanol ligated to the catalytic zinc in the classical tetrahedral coordination with Cys-43, Cys-153, and His-66. In contrast, the B and D subunits have an open conformation with no bound coenzyme, and the catalytic zinc has an alternative, inverted coordination with Cys-43, Cys-153, His-66, and the carboxylate of Glu-67. The asymmetry in the dimeric subunits of the tetramer provides two structures that appear to be relevant for the catalytic mechanism. The alternative coordination of the zinc may represent an intermediate in the mechanism of displacement of the zinc-bound water with alcohol or aldehyde substrates. Substitution of Glu-67 with Gln-67 decreases the catalytic efficiency by 100-fold. Previous studies of structural modeling, evolutionary relationships, substrate specificity, chemical modification, and site-directed mutagenesis are interpreted more fully with the three-dimensional structure. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7kcb.cif.gz | 239 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7kcb.ent.gz | 191.5 KB | Display | PDB format |
| PDBx/mmJSON format | 7kcb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kc/7kcbftp://data.pdbj.org/pub/pdb/validation_reports/kc/7kcb | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  22807MC  7kc2C  7kcqC  7kjyC C: citing same article ( M: map data used to model this data |
|---|---|
| Similar structure data |















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 36759.906 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Source: (natural) #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Zn / Feature type: SUBJECT OF INVESTIGATION Mass: 65.409 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Zn / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | ChemComp-NAD /   Mass: 663.425 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Feature type: SUBJECT OF INVESTIGATION / Comment: NAD*YM Mass: 663.425 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Feature type: SUBJECT OF INVESTIGATION / Comment: NAD*YM#4: Chemical | ChemComp-ETF / 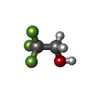  Mass: 100.040 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H3F3O / Feature type: SUBJECT OF INVESTIGATION Mass: 100.040 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H3F3O / Feature type: SUBJECT OF INVESTIGATIONHas ligand of interest | Y | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: Alcohol Dehydrogenase NAD+ Pyrazole complex / Type: COMPLEX / Entity ID: #1 / Source: NATURAL |
|---|---|
| Molecular weight | Value: 0.37 MDa / Experimental value: NO |
| Source (natural) | Organism: |
| Buffer solution | pH: 8.2 Details: Tris HCl buffer 5mM with 200mM KCl adjusted to pH 8.2. |
| Buffer component | Conc.: 50 mM / Name: Tris / Formula: C4H11NO3 |
| Specimen | Conc.: 5 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES / Details: Purified by Size Exclusion chromatography |
| Specimen support | Grid material: GOLD / Grid mesh size: 300 divisions/in. / Grid type: Quantifoil |
| Vitrification | Instrument: FEI VITROBOT MARK II / Cryogen name: ETHANE / Humidity: 100 % / Chamber temperature: 298 K |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: FEI TITAN KRIOS |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: OTHER FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: OTHER |
| Electron lens | Mode: BRIGHT FIELD / Nominal defocus max: 2000 nm / Nominal defocus min: 800 nm / Cs: 2.7 mm / C2 aperture diameter: 100 µm |
| Specimen holder | Cryogen: NITROGEN |
| Image recording | Electron dose: 54 e/Å2 / Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) |
- Processing
Processing
| EM software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: NONE | ||||||||||||||||||||||||
| 3D reconstruction | Resolution: 2.77 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 1284904 / Algorithm: BACK PROJECTION / Symmetry type: POINT | ||||||||||||||||||||||||
| Atomic model building | B value: 39.3 / Protocol: RIGID BODY FIT / Space: REAL / Target criteria: Correlation Coefficient | ||||||||||||||||||||||||
| Atomic model building | PDB-ID: 5ENV Pdb chain-ID: A / Accession code: 5ENV / Pdb chain residue range: 1-347 / Source name: PDB / Type: experimental model | ||||||||||||||||||||||||
| Refinement | Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2 | ||||||||||||||||||||||||
| Displacement parameters | Biso mean: 43.6 Å2 | ||||||||||||||||||||||||
| Refine LS restraints |
|
