 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7dlm | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Short chain dehydrogenase (SCR) crystal structure with NADPH | |||||||||
 Components Components | Carbonyl Reductase | |||||||||
 Keywords Keywords | OXIDOREDUCTASE / Rossman fold / tetramer / tag-free / wild type with NADPH | |||||||||
| Function / homology |  Function and homology information Function and homology informationmannitol 2-dehydrogenase (NADP+) activity / oxidoreductase activity, acting on NAD(P)H, oxygen as acceptor / small molecule metabolic process / Oxidoreductases; Acting on the CH-OH group of donors; With NAD+ or NADP+ as acceptor / carbohydrate metabolic process Similarity search - Function | |||||||||
| Biological species |  Candida parapsilosis (fungus) Candida parapsilosis (fungus) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.59 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.59 Å | |||||||||
 Authors Authors | Li, Y.H. / Zhang, R.Z. / Forouhar, F. / Wang, C. / Montelione, G.T. / Szyperski, T. / Xu, Y. / Hunt, J.F. | |||||||||
| Funding support |  China, 2items China, 2items
| |||||||||
 Citation Citation | Journal: EMBO J / Year: 2022 Title: Oligomeric interactions maintain active-site structure in a noncooperative enzyme family. Authors: Yaohui Li / Rongzhen Zhang / Chi Wang / Farhad Forouhar / Oliver B Clarke / Sergey Vorobiev / Shikha Singh / Gaetano T Montelione / Thomas Szyperski / Yan Xu / John F Hunt /  Abstract: The evolutionary benefit accounting for widespread conservation of oligomeric structures in proteins lacking evidence of intersubunit cooperativity remains unclear. Here, crystal and cryo-EM ...The evolutionary benefit accounting for widespread conservation of oligomeric structures in proteins lacking evidence of intersubunit cooperativity remains unclear. Here, crystal and cryo-EM structures, and enzymological data, demonstrate that a conserved tetramer interface maintains the active-site structure in one such class of proteins, the short-chain dehydrogenase/reductase (SDR) superfamily. Phylogenetic comparisons support a significantly longer polypeptide being required to maintain an equivalent active-site structure in the context of a single subunit. Oligomerization therefore enhances evolutionary fitness by reducing the metabolic cost of enzyme biosynthesis. The large surface area of the structure-stabilizing oligomeric interface yields a synergistic gain in fitness by increasing tolerance to activity-enhancing yet destabilizing mutations. We demonstrate that two paralogous SDR superfamily enzymes with different specificities can form mixed heterotetramers that combine their individual enzymological properties. This suggests that oligomerization can also diversify the functions generated by a given metabolic investment, enhancing the fitness advantage provided by this architectural strategy. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7dlm.cif.gz | 161.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7dlm.ent.gz | 101.2 KB | Display | PDB format |
| PDBx/mmJSON format | 7dlm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dl/7dlmftp://data.pdbj.org/pub/pdb/validation_reports/dl/7dlm | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7dldC  7dllC  7dmgC  7dn1C  7vyqC  3ctmS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |



-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 30204.873 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Candida parapsilosis (fungus) / Gene: DQ675534 / Production host:  References: UniProt: B2KJ46, Oxidoreductases; Acting on the CH-OH group of donors; With NAD+ or NADP+ as acceptor #2: Chemical | 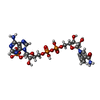  Mass: 745.421 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H30N7O17P3 / Feature type: SUBJECT OF INVESTIGATION Mass: 745.421 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H30N7O17P3 / Feature type: SUBJECT OF INVESTIGATION#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 483 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 483 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.09 Å3/Da / Density % sol: 41.13 % |
|---|---|
| Crystal grow | Temperature: 293.15 K / Method: microbatch / pH: 7 / Details: 200 mM Ammonium Chloride, 20% PEG 3350, pH 7.0 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS-II / Beamline: 19-ID / Wavelength: 0.9795 Å |
| Detector | Type: ADSC HF-4M / Detector: PIXEL / Date: Aug 20, 2018 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 1.59→29 Å / Num. obs: 67312 / % possible obs: 99.1 % / Redundancy: 9.3 % / Biso Wilson estimate: 16 Å2 / CC1/2: 0.999 / Net I/σ(I): 19.6 |
| Reflection shell | Resolution: 1.59→1.63 Å / Num. unique obs: 4630 / CC1/2: 0.696 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3CTM Resolution: 1.59→28.99 Å / SU ML: 0.1674 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 18.6314 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.11 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.59→28.99 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
