 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7ct9 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of SAH bound CmoB from Vibrio Vulnificus | ||||||
 Components Components | tRNA U34 carboxymethyltransferase | ||||||
 Keywords Keywords | TRANSFERASE / carboxymethyl transferase / tRNA modification / SAH / S-adenosyl homocysteine | ||||||
| Function / homology |  Function and homology information Function and homology informationtRNA wobble uridine modification / Transferases; Transferring alkyl or aryl groups, other than methyl groups / transferase activity, transferring alkyl or aryl (other than methyl) groups / methyltransferase activity Similarity search - Function | ||||||
| Biological species |  Vibrio vulnificus MO6-24/O (bacteria) Vibrio vulnificus MO6-24/O (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Kim, J. / Jeong, S. | ||||||
| Funding support |  Korea, Republic Of, 1items Korea, Republic Of, 1items
| ||||||
 Citation Citation | Journal: Biochem.Biophys.Res.Commun. / Year: 2021 Title: Structural snapshots of CmoB in various states during wobble uridine modification of tRNA. Authors: Jeong, S. / Kim, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7ct9.cif.gz | 156.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7ct9.ent.gz | 119.3 KB | Display | PDB format |
| PDBx/mmJSON format | 7ct9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ct/7ct9ftp://data.pdbj.org/pub/pdb/validation_reports/ct/7ct9 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7ct8C  7ctaC  4qnxS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: _ / Ens-ID: 1 / Beg auth comp-ID: MET / Beg label comp-ID: MET / End auth comp-ID: SAH / End label comp-ID: SAH / Refine code: _ / Auth seq-ID: 0 - 401 / Label seq-ID: 1
|
-Components
| #1: Protein | Mass: 38393.793 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Vibrio vulnificus MO6-24/O (bacteria) / Strain: MO6-24/OGene: cmoB, CRN46_03385, CRN52_23395, D8T49_20765, D8T54_03695, JS86_05215 Production host: References: UniProt: A0A087JHK9, UniProt: Q8DAP0*PLUS, Transferases; Transferring alkyl or aryl groups, other than methyl groups #2: Chemical |   Type: L-peptide linking / Mass: 384.411 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H20N6O5S / Feature type: SUBJECT OF INVESTIGATION Type: L-peptide linking / Mass: 384.411 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H20N6O5S / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | 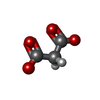  Mass: 102.046 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H2O4 Mass: 102.046 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H2O4#4: Chemical |   Mass: 94.971 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: PO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 386 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 386 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.67 Å3/Da / Density % sol: 53.92 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / Details: Sodium malonate, PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PAL/PLS / Beamline: 7A (6B, 6C1) / Wavelength: 0.97919 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 270 / Detector: CCD / Date: Mar 13, 2019 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97919 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.3→50 Å / Num. obs: 36969 / % possible obs: 99.9 % / Redundancy: 4 % / Rmerge(I) obs: 0.119 / Rpim(I) all: 0.069 / Rrim(I) all: 0.138 / Χ2: 1.636 / Net I/σ(I): 6.8 / Num. measured all: 147619 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4qnx Resolution: 2.3→30.41 Å / Cor.coef. Fo:Fc: 0.961 / Cor.coef. Fo:Fc free: 0.932 / SU B: 7.749 / SU ML: 0.178 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.289 / ESU R Free: 0.219 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 101.89 Å2 / Biso mean: 25.242 Å2 / Biso min: 10.39 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.3→30.41 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Ens-ID: 1 / Number: 11046 / Refine-ID: X-RAY DIFFRACTION / Type: interatomic distance / Rms dev position: 0.07 Å / Weight position: 0.05
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.36 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
