 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7bts: Structure of human beta1 adrenergic receptor bound to epinephrine... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7bts | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of human beta1 adrenergic receptor bound to epinephrine and nanobody 6B9 | ||||||
 Components Components |
| ||||||
 Keywords Keywords | MEMBRANE PROTEIN / G protein coupled receptor | ||||||
| Function / homology |  Function and homology information Function and homology informationviral release from host cell by cytolysis / peptidoglycan catabolic process / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / host cell cytoplasm / defense response to bacterium Similarity search - Function | ||||||
| Biological species |  Enterobacteria phage T4 (virus) Enterobacteria phage T4 (virus) Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.13 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.13 Å | ||||||
 Authors Authors | Xu, X. / Kaindl, J. / Clark, M. / Hubner, H. / Hirata, K. / Sunahara, R. / Gmeiner, P. / Kobilka, B.K. / Liu, X. | ||||||
 Citation Citation | Journal: Cell Res. / Year: 2021 Title: Binding pathway determines norepinephrine selectivity for the human beta 1 AR over beta 2 AR. Authors: Xu, X. / Kaindl, J. / Clark, M.J. / Hubner, H. / Hirata, K. / Sunahara, R.K. / Gmeiner, P. / Kobilka, B.K. / Liu, X. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7bts.cif.gz | 141 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7bts.ent.gz | 97.1 KB | Display | PDB format |
| PDBx/mmJSON format | 7bts.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bt/7btsftp://data.pdbj.org/pub/pdb/validation_reports/bt/7bts | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7bu6C  7bu7C  7bvqC  4ldeS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
-Protein / Antibody , 2 types, 2 molecules AB
| #1: Protein | Mass: 52235.879 Da / Num. of mol.: 1 / Mutation: C944T,C987A Source method: isolated from a genetically manipulated source Details: Chain A 1261 Cys to Chain A 1314 LEU are truncated region. Source: (gene. exp.) Enterobacteria phage T4 (virus), (gene. exp.) Homo sapiens (human)Gene: e, T4Tp126, ADRB1, ADRB1R, B1AR / Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: D9IEF7, lysozyme Spodoptera frugiperda (fall armyworm) / References: UniProt: D9IEF7, lysozyme |
|---|---|
| #2: Antibody | Mass: 12949.375 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Spodoptera frugiperda (fall armyworm) |
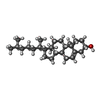
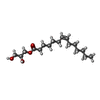
-Non-polymers , 6 types, 11 molecules 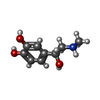



| #3: Chemical | ChemComp-ALE /  Mass: 183.204 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H13NO3 / Feature type: SUBJECT OF INVESTIGATION / Comment: medication, hormone*YM Mass: 183.204 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H13NO3 / Feature type: SUBJECT OF INVESTIGATION / Comment: medication, hormone*YM | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #4: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: SO4#5: Chemical | ChemComp-NA / |  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na#6: Chemical | ChemComp-EPE / |  Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM#7: Chemical | ChemComp-CLR / |  Mass: 386.654 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H46O Mass: 386.654 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H46O#8: Chemical | ChemComp-1WV / ( |  Mass: 300.434 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H32O4 Mass: 300.434 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H32O4 |
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | Y |
| Sequence details | THE GENEBANK ENTRY NP_000675 IS A REFERENCE SEQUENCE FOR THE RESIDUES FROM 171TH to 462TH OF CHAIN A. |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.59 Å3/Da / Density % sol: 73.2 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: lipidic cubic phase Details: 100 mM HEPES, pH 7.5, 80-180 mM Sodium Sulfate, 38-42% PEG400 or PEG300, 10mM epinephrine |
-Data collection
| Diffraction | Mean temperature: 80 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL32XU / Wavelength: 1 Å / Beamline: BL32XU / Wavelength: 1 Å |
| Detector | Type: DECTRIS EIGER X 9M / Detector: PIXEL / Date: Feb 4, 2018 |
| Radiation | Monochromator: liquid nitrogen-cooled double crystal Si(111) Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 3.1→50 Å / Num. obs: 20331 / % possible obs: 96.8 % / Redundancy: 11.4 % / Biso Wilson estimate: 74.88 Å2 / CC1/2: 0.982 / Net I/σ(I): 5.59 |
| Reflection shell | Resolution: 3.1→3.2 Å / Redundancy: 7.6 % / Mean I/σ(I) obs: 1.37 / Num. unique obs: 897 / CC1/2: 0.5 / % possible all: 58.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4LDE Resolution: 3.13→19.98 Å / SU ML: 0.429 / Cross valid method: FREE R-VALUE / σ(F): 1.36 / Phase error: 26.9635 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 80.32 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.13→19.98 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
