 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7br4: Structure of deletion mutant of alpha-glucuronidase (TM0752) from... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7br4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of deletion mutant of alpha-glucuronidase (TM0752) from Thermotoga maritima | ||||||
 Components Components | Alpha-glucosidase, putative | ||||||
 Keywords Keywords | HYDROLASE / hydrolase-oxidoreductase / alpha-glucuronidase activity / carbohydrate metabolism / NAD-binding Rossmann-fold / LDH C-terminal domain-like | ||||||
| Function / homology |  Function and homology information Function and homology informationoxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor / hydrolase activity, hydrolyzing O-glycosyl compounds / carbohydrate metabolic process / nucleotide binding / metal ion binding / cytosol Similarity search - Function | ||||||
| Biological species |   Thermotoga maritima (bacteria) Thermotoga maritima (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.95 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.95 Å | ||||||
 Authors Authors | Manoj, N. / Mohapatra, S.B. | ||||||
 Citation Citation | Journal: Biochim Biophys Acta Proteins Proteom / Year: 2021 Title: A conserved pi-helix plays a key role in thermoadaptation of catalysis in the glycoside hydrolase family 4. Authors: Mohapatra, S.B. / Manoj, N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7br4.cif.gz | 287.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7br4.ent.gz | 232.4 KB | Display | PDB format |
| PDBx/mmJSON format | 7br4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 7br4_validation.pdf.gz | 759.7 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 7br4_full_validation.pdf.gz | 761.4 KB | Display | |
| Data in XML | 7br4_validation.xml.gz | 21 KB | Display | |
| Data in CIF | 7br4_validation.cif.gz | 31 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/br/7br4ftp://data.pdbj.org/pub/pdb/validation_reports/br/7br4 | HTTPS FTP |
-Related structure data
| Related structure data |  1vjtS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 56748.676 Da / Num. of mol.: 1 / Mutation: Deletion Phe407 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermotoga maritima (strain ATCC 43589 / MSB8 / DSM 3109 / JCM 10099) (bacteria)Strain: ATCC 43589 / MSB8 / DSM 3109 / JCM 10099 / Gene: TM_0752 / Plasmid: pMH2T7 / Production host: |
|---|---|
| #2: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn |
| #3: Chemical | ChemComp-NAI / 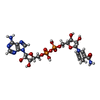  Mass: 665.441 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H29N7O14P2 / Feature type: SUBJECT OF INVESTIGATION Mass: 665.441 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H29N7O14P2 / Feature type: SUBJECT OF INVESTIGATION |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 299 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 299 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.26 Å3/Da / Density % sol: 45.6 % |
|---|---|
| Crystal grow | Temperature: 300 K / Method: vapor diffusion, hanging drop / pH: 6 Details: 12% PEG 3350, 0.2 M trilithium citrate, 0.1 M imidazole pH 6.0, 2-propanol PH range: 5.8 - 6.2 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: BRUKER AXS MICROSTAR / Wavelength: 1.5418 Å | |||||||||||||||||||||||||||
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Jul 19, 2017 | |||||||||||||||||||||||||||
| Radiation | Monochromator: Double mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 | |||||||||||||||||||||||||||
| Reflection | Resolution: 1.95→43.45 Å / Num. obs: 36415 / % possible obs: 96.5 % / Redundancy: 4.3 % / Biso Wilson estimate: 20.39 Å2 / CC1/2: 0.992 / Rmerge(I) obs: 0.117 / Rpim(I) all: 0.061 / Rrim(I) all: 0.132 / Net I/σ(I): 6.2 / Num. measured all: 156648 / Scaling rejects: 179 | |||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1VJT Resolution: 1.95→29.46 Å / SU ML: 0.24 / Cross valid method: THROUGHOUT / σ(F): 1.35 / Phase error: 24.17
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 84.31 Å2 / Biso mean: 27.2354 Å2 / Biso min: 10.33 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.95→29.46 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
