| 登録情報 | データベース: PDB / ID: 7b0e
|
|---|
| タイトル | Crystal structure of SmbA loop deletion mutant |
|---|
 要素 要素 | SmbA |
|---|
 キーワード キーワード | SIGNALING PROTEIN / c-di-GMP receptor / ppGpp receptor / second messenger / TIM barrel |
|---|
| 機能・相同性 | NADP-dependent oxidoreductase domain / NADP-dependent oxidoreductase domain superfamily / nucleotide binding / TIM Barrel / Alpha-Beta Barrel / metal ion binding / Alpha Beta / Chem-C2E / NADP-dependent oxidoreductase domain-containing protein 機能・相同性情報 機能・相同性情報 |
|---|
| 生物種 |  Caulobacter vibrioides CB15 (バクテリア) Caulobacter vibrioides CB15 (バクテリア) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.4 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.4 Å |
|---|
 データ登録者 データ登録者 | Dubey, B.N. / Schirmer, T. |
|---|
| 資金援助 |  スイス, 1件 スイス, 1件 | 組織 | 認可番号 | 国 |
|---|
| Swiss National Science Foundation | 31003A_166652 | スイス |
|
|---|
 引用 引用 | ジャーナル: To Be Published
タイトル: High-resolution crystal structure of loop deletion mutant SmbA bound to a monomeric c-di-GMP
著者: Dubey, B.N. / Shyp, V. / Jenal, U. / Schirmer, T. |
|---|
| 履歴 | | 登録 | 2020年11月19日 | 登録サイト: PDBE / 処理サイト: PDBE |
|---|
| 改定 1.0 | 2021年12月1日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 2.0 | 2022年9月7日 | Group: Advisory / Atomic model ...Advisory / Atomic model / Data collection / Derived calculations / Non-polymer description / Refinement description / Source and taxonomy / Structure summary
カテゴリ: atom_site / atom_site_anisotrop ...atom_site / atom_site_anisotrop / atom_type / chem_comp / entity / entity_src_gen / pdbx_contact_author / pdbx_distant_solvent_atoms / pdbx_entity_nonpoly / pdbx_nonpoly_scheme / pdbx_struct_assembly_gen / pdbx_struct_conn_angle / pdbx_validate_close_contact / pdbx_validate_torsion / refine / refine_hist / refine_ls_restr / refine_ls_shell / software / struct_asym / struct_conn / struct_site / struct_site_gen
Item: _atom_site_anisotrop.U[1][1] / _atom_site_anisotrop.U[1][2] ..._atom_site_anisotrop.U[1][1] / _atom_site_anisotrop.U[1][2] / _atom_site_anisotrop.U[1][3] / _atom_site_anisotrop.U[2][2] / _atom_site_anisotrop.U[2][3] / _atom_site_anisotrop.U[3][3] / _chem_comp.formula / _chem_comp.formula_weight / _chem_comp.id / _chem_comp.mon_nstd_flag / _chem_comp.name / _chem_comp.pdbx_synonyms / _chem_comp.type / _entity_src_gen.pdbx_gene_src_scientific_name / _pdbx_struct_assembly_gen.asym_id_list / _pdbx_validate_torsion.phi / _pdbx_validate_torsion.psi / _refine.B_iso_max / _refine.B_iso_mean / _refine.B_iso_min / _refine.ls_R_factor_R_free / _refine.ls_R_factor_R_work / _refine.ls_R_factor_obs / _refine.overall_SU_ML / _refine.pdbx_overall_phase_error / _refine_hist.number_atoms_solvent / _refine_hist.number_atoms_total / _refine_hist.pdbx_B_iso_mean_ligand / _refine_hist.pdbx_B_iso_mean_solvent / _refine_hist.pdbx_number_atoms_ligand / _refine_ls_restr.dev_ideal / _refine_ls_restr.number / _refine_ls_shell.R_factor_R_free / _refine_ls_shell.R_factor_R_work / _refine_ls_shell.d_res_high / _refine_ls_shell.d_res_low / _refine_ls_shell.number_reflns_R_work / _software.classification / _software.name / _software.version
解説: Occupancy of atoms on special symmetry positions / Provider: author / タイプ: Coordinate replacement |
|---|
| 改定 2.1 | 2024年1月31日 | Group: Data collection / Refinement description
カテゴリ: chem_comp_atom / chem_comp_bond / pdbx_initial_refinement_model |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード










 PDBj
PDBj

 集合体
集合体
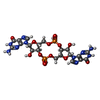




 分子量: 690.411 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C20H24N10O14P2 / タイプ: SUBJECT OF INVESTIGATION
分子量: 690.411 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C20H24N10O14P2 / タイプ: SUBJECT OF INVESTIGATION 分子量: 62.068 Da / 分子数: 12 / 由来タイプ: 合成 / 式: C2H6O2
分子量: 62.068 Da / 分子数: 12 / 由来タイプ: 合成 / 式: C2H6O2 分子量: 22.990 Da / 分子数: 2 / 由来タイプ: 天然 / 式: Na
分子量: 22.990 Da / 分子数: 2 / 由来タイプ: 天然 / 式: Na 分子量: 40.078 Da / 分子数: 3 / 由来タイプ: 合成 / 式: Ca
分子量: 40.078 Da / 分子数: 3 / 由来タイプ: 合成 / 式: Ca 試料調製
試料調製 解析
解析