 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6xtv: FULL-LENGTH LTTR LYSG FROM CORYNEBACTERIUM GLUTAMICUM WITH BOUND ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6xtv | ||||||
|---|---|---|---|---|---|---|---|
| Title | FULL-LENGTH LTTR LYSG FROM CORYNEBACTERIUM GLUTAMICUM WITH BOUND EFFECTOR ARG | ||||||
 Components Components | Lysine export transcriptional regulatory protein LysG | ||||||
 Keywords Keywords | TRANSCRIPTION / LTTR HELIX-TURN-HELIX TRANSCRIPTION REGULATION | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA-binding transcription factor activity / DNA-templated transcription / DNA binding Similarity search - Function | ||||||
| Biological species |  Corynebacterium glutamicum MB001 (bacteria) Corynebacterium glutamicum MB001 (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 3.3 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 3.3 Å | ||||||
 Authors Authors | Hofmann, E. / Syberg, F. / Schlicker, C. / Eggeling, L. / Schendzielorz, G. | ||||||
| Funding support |  Germany, 1items Germany, 1items
| ||||||
 Citation Citation | Journal: Nat Commun / Year: 2020 Title: Engineering and application of a biosensor with focused ligand specificity. Authors: Della Corte, D. / van Beek, H.L. / Syberg, F. / Schallmey, M. / Tobola, F. / Cormann, K.U. / Schlicker, C. / Baumann, P.T. / Krumbach, K. / Sokolowsky, S. / Morris, C.J. / Grunberger, A. / ...Authors: Della Corte, D. / van Beek, H.L. / Syberg, F. / Schallmey, M. / Tobola, F. / Cormann, K.U. / Schlicker, C. / Baumann, P.T. / Krumbach, K. / Sokolowsky, S. / Morris, C.J. / Grunberger, A. / Hofmann, E. / Schroder, G.F. / Marienhagen, J. #1: Journal: Acta Crystallogr., Sect. D: Biol. Crystallogr. / Year: 2012Title: Towards automated crystallographic structure refinement with phenix.refine. Authors: Afonine, P.V. / Grosse-Kunstleve, R.W. / Echols, N. / Headd, J.J. / Moriaty, N.W. / Mustyakimov, M. / Terwilliger, T.C. / Urzhumtsev, A. / Zwart, P.H. / Adams, P.D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6xtv.cif.gz | 338.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6xtv.ent.gz | 274.3 KB | Display | PDB format |
| PDBx/mmJSON format | 6xtv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xt/6xtvftp://data.pdbj.org/pub/pdb/validation_reports/xt/6xtv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6xtuSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 31423.100 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Corynebacterium glutamicum MB001 (bacteria)Gene: lysG, Cgl1263, cg1425 / Production host: #2: Chemical | ChemComp-ARG / | 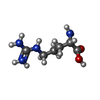  Type: L-peptide linking / Mass: 175.209 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15N4O2 / Feature type: SUBJECT OF INVESTIGATION Type: L-peptide linking / Mass: 175.209 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15N4O2 / Feature type: SUBJECT OF INVESTIGATIONHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.32 Å3/Da / Density % sol: 47 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 8 Details: 0.1 M SODIUM CHLORIDE; 0.1 M TRIS PH REMARK 280 8.5, 30 %(V/V) PEG 400 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM30A / Wavelength: 0.979657 Å / Beamline: BM30A / Wavelength: 0.979657 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Feb 22, 2013 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979657 Å / Relative weight: 1 |
| Reflection | Resolution: 3→47 Å / Num. obs: 17846 / % possible obs: 99.65 % / Redundancy: 7.2 % / Biso Wilson estimate: 86.96 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.1299 / Rpim(I) all: 0.05121 / Rrim(I) all: 0.1399 / Net I/σ(I): 15.77 |
| Reflection shell | Resolution: 3→3.111 Å / Rmerge(I) obs: 1.94 / Mean I/σ(I) obs: 1.15 / Num. unique obs: 12966 / CC1/2: 0.428 / Rpim(I) all: 0.7546 / Rrim(I) all: 2.084 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: 6XTU Resolution: 3.3→47 Å / Cross valid method: FREE R-VALUE
| ||||||||||||||||||||||||
| Displacement parameters | Biso mean: 92.26 Å2 | ||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.3→47 Å
| ||||||||||||||||||||||||
| Refine LS restraints |
|
