 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6xs3: X-ray structure of the monoclinic crystal form at 2.48 A resoluti... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6xs3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray structure of the monoclinic crystal form at 2.48 A resolution of lipase from Thermomyces (Humicola) lanuginosa at 298 K | ||||||
 Components Components | Lipase | ||||||
 Keywords Keywords | LIPID BINDING PROTEIN / substrate complex / diacylglycerol / covalent intermediate / interfacial activation | ||||||
| Function / homology |  Function and homology information Function and homology informationtriacylglycerol lipase / triacylglycerol lipase activity / lipid catabolic process Similarity search - Function | ||||||
| Biological species |   Thermomyces lanuginosus (fungus) Thermomyces lanuginosus (fungus) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.48 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.48 Å | ||||||
 Authors Authors | McPherson, A. | ||||||
 Citation Citation | Journal: Current Enzyme Inhibition / Year: 2020 Title: The crystal Structures of Thermomyces (Humicola) lanuginosa lipase in complex with enzymatic reactants Authors: McPherson, A. / Larson, S.B. / Kalasky, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6xs3.cif.gz | 597.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6xs3.ent.gz | 501.3 KB | Display | PDB format |
| PDBx/mmJSON format | 6xs3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xs/6xs3ftp://data.pdbj.org/pub/pdb/validation_reports/xs/6xs3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6or3C  6xokC  6xrvC  4ea6S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein / Sugars , 2 types, 12 molecules ABCEDF
| #1: Protein | Mass: 31836.459 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermomyces lanuginosus (fungus) / Gene: LIP / Production host: #4: Sugar | ChemComp-NAG /  Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 6 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 6Source method: isolated from a genetically manipulated source Formula: C8H15NO6 |
|---|
-Non-polymers , 5 types, 453 molecules 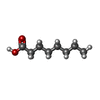



| #2: Chemical | ChemComp-OCA /  Mass: 144.211 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C8H16O2 Mass: 144.211 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C8H16O2#3: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Ca#5: Chemical | ChemComp-LTV /  Mass: 723.204 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C46H90O5 / Feature type: SUBJECT OF INVESTIGATION Mass: 723.204 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C46H90O5 / Feature type: SUBJECT OF INVESTIGATION#6: Chemical | ChemComp-PO4 /  Mass: 94.971 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: PO4#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 428 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.32 Å3/Da / Density % sol: 46.91 % / Description: cube shaped blocks |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: CXrystals were grown by sitting drop in Cryschem plates using 0.6 ml reservoirs of 20% PEG 3350 in 0.1M MES buffer. The drops were 6 ul composed of equal amounts of protein at 30 mg/ml in ...Details: CXrystals were grown by sitting drop in Cryschem plates using 0.6 ml reservoirs of 20% PEG 3350 in 0.1M MES buffer. The drops were 6 ul composed of equal amounts of protein at 30 mg/ml in water with the reservoir solution. Temperature was 298 K and crystals appeared after about two weeks PH range: 6.0 - 7.0 |
-Data collection
| Diffraction | Mean temperature: 298 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.5419 Å |
| Detector | Type: OXFORD RUBY CCD / Detector: CCD / Date: Jun 20, 2017 |
| Radiation | Monochromator: Osmic mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5419 Å / Relative weight: 1 |
| Reflection | Resolution: 2.48→75 Å / Num. obs: 81074 / % possible obs: 100 % / Redundancy: 9.6 % / Biso Wilson estimate: 26 Å2 / CC1/2: 0.979 / Rmerge(I) obs: 0.292 / Rpim(I) all: 0.102 / Rrim(I) all: 0.327 / Rsym value: 0.287 / Net I/σ(I): 7.1 |
| Reflection shell | Resolution: 2.48→2.55 Å / Redundancy: 3.9 % / Rmerge(I) obs: 1.02 / Mean I/σ(I) obs: 1.3 / Num. unique obs: 4362 / CC1/2: 0.18 / Rpim(I) all: 0.62 / Rrim(I) all: 1.3 / Rsym value: 1.01 / % possible all: 96.2 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4EA6 Resolution: 2.48→51.27 Å / SU ML: 0.32 / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 21.19 / Stereochemistry target values: ML Details: Structure was refined to convergence with REFMAC5 in CCP4, then five runs through PHENIX REFINE using isotropic B values and no TLS.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 164 Å2 / Biso mean: 36.1154 Å2 / Biso min: 9.62 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.48→51.27 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 22
|
