 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6xeg: Crystal structure of the PTP1B YopH WPD loop Chimera 4 bound to t... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6xeg | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the PTP1B YopH WPD loop Chimera 4 bound to tungstate | ||||||
 Components Components | Tyrosine-protein phosphatase non-receptor type 1 | ||||||
 Keywords Keywords | HYDROLASE / PTP1B / tyrosine phosphatase / WPD loop | ||||||
| Function / homology |  Function and homology information Function and homology informationPTK6 Down-Regulation / regulation of hepatocyte growth factor receptor signaling pathway / positive regulation of receptor catabolic process / peptidyl-tyrosine dephosphorylation involved in inactivation of protein kinase activity / insulin receptor recycling / negative regulation of vascular endothelial growth factor receptor signaling pathway / positive regulation of IRE1-mediated unfolded protein response / negative regulation of PERK-mediated unfolded protein response / IRE1-mediated unfolded protein response / regulation of intracellular protein transport ...PTK6 Down-Regulation / regulation of hepatocyte growth factor receptor signaling pathway / positive regulation of receptor catabolic process / peptidyl-tyrosine dephosphorylation involved in inactivation of protein kinase activity / insulin receptor recycling / negative regulation of vascular endothelial growth factor receptor signaling pathway / positive regulation of IRE1-mediated unfolded protein response / negative regulation of PERK-mediated unfolded protein response / IRE1-mediated unfolded protein response / regulation of intracellular protein transport / cytoplasmic side of endoplasmic reticulum membrane / sorting endosome / mitochondrial crista / platelet-derived growth factor receptor-beta signaling pathway / regulation of type I interferon-mediated signaling pathway / regulation of endocytosis / positive regulation of protein tyrosine kinase activity / non-membrane spanning protein tyrosine phosphatase activity / peptidyl-tyrosine dephosphorylation / Regulation of IFNA/IFNB signaling / cellular response to unfolded protein / regulation of signal transduction / growth hormone receptor signaling pathway via JAK-STAT / negative regulation of endoplasmic reticulum stress-induced intrinsic apoptotic signaling pathway / negative regulation of signal transduction / Regulation of IFNG signaling / Growth hormone receptor signaling / MECP2 regulates neuronal receptors and channels / negative regulation of MAP kinase activity / endoplasmic reticulum unfolded protein response / positive regulation of JUN kinase activity / negative regulation of insulin receptor signaling pathway / Insulin receptor recycling / protein dephosphorylation / ephrin receptor binding / Integrin signaling / protein-tyrosine-phosphatase / protein tyrosine phosphatase activity / protein phosphatase 2A binding / endosome lumen / insulin receptor binding / Negative regulation of MET activity / negative regulation of ERK1 and ERK2 cascade / receptor tyrosine kinase binding / insulin receptor signaling pathway / actin cytoskeleton organization / early endosome / mitochondrial matrix / cadherin binding / protein kinase binding / enzyme binding / endoplasmic reticulum / protein-containing complex / RNA binding / zinc ion binding / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.549 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.549 Å | ||||||
 Authors Authors | Olsen, K.J. / Shen, R. / Johnson, S.J. / Hengge, A.C. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Chem Sci / Year: 2022 Title: Insights into the importance of WPD-loop sequence for activity and structure in protein tyrosine phosphatases. Authors: Shen, R. / Crean, R.M. / Olsen, K.J. / Corbella, M. / Calixto, A.R. / Richan, T. / Brandao, T.A.S. / Berry, R.D. / Tolman, A. / Loria, J.P. / Johnson, S.J. / Kamerlin, S.C.L. / Hengge, A.C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6xeg.cif.gz | 142.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6xeg.ent.gz | 109.1 KB | Display | PDB format |
| PDBx/mmJSON format | 6xeg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 6xeg_validation.pdf.gz | 464.3 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 6xeg_full_validation.pdf.gz | 466.3 KB | Display | |
| Data in XML | 6xeg_validation.xml.gz | 13.9 KB | Display | |
| Data in CIF | 6xeg_validation.cif.gz | 18.6 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xe/6xegftp://data.pdbj.org/pub/pdb/validation_reports/xe/6xeg | HTTPS FTP |
-Related structure data
| Related structure data |  6xe8C  6xeaC  6xedC  6xeeC  6xefC  7s4fC  3qkpS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj

















- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 37375.590 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PTPN1, PTP1B / Production host:  |
|---|
-Non-polymers , 5 types, 93 molecules 
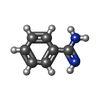



| #2: Chemical | ChemComp-TRS /  Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM |
|---|---|
| #3: Chemical | ChemComp-BEN /  Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 |
| #4: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #5: Chemical | ChemComp-WO4 /  Mass: 247.838 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: WO4 Mass: 247.838 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: WO4 |
| #6: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 89 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has ligand of interest | N |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.18 Å3/Da / Density % sol: 61.28 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 7.8 Details: tris hydrochloride pH 7.8, magnesium acetate tetrahydrate, PEG 8000 and benzamidine PH range: 7.8-8.5 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL / Beamline: BL9-1 / Wavelength: 0.97946 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Jan 31, 2019 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97946 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.549→50 Å / Num. obs: 15928 / % possible obs: 99.6 % / Redundancy: 16.2 % / Rmerge(I) obs: 0.155 / Rpim(I) all: 0.038 / Rrim(I) all: 0.16 / Χ2: 0.707 / Net I/σ(I): 3.7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3QKP Resolution: 2.549→44.322 Å / SU ML: 0.22 / Cross valid method: THROUGHOUT / σ(F): 1.36 / Phase error: 18.56 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 102.91 Å2 / Biso mean: 47.2032 Å2 / Biso min: 20.31 Å2 | ||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.549→44.322 Å
| ||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0
| ||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -44.5124 Å / Origin y: 17.1428 Å / Origin z: 15.4687 Å
| ||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
