Entry Database : PDB / ID : 1nweTitle Ptp1B R47C Modified at C47 with N-[4-(2-{2-[3-(2-Bromo-acetylamino)-propionylamino]-3-hydroxy-propionylamino}-ethyl)-phenyl]-oxalamic acid Protein-tyrosine phosphatase, non-receptor type 1 Keywords / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / Resolution : 3.1 Å Authors Erlanson, D.A. / McDowell, R.S. / He, M.M. / Randal, M. / Simmons, R.L. / Kung, J. / Waight, A. / Hansen, S.K. Journal : J.Am.Chem.Soc. / Year : 2003Title : Discovery of a New Phosphotyrosine Mimetic for PTP1B Using Breakaway TetheringAuthors : Erlanson, D.A. / McDowell, R.S. / He, M.M. / Randal, M. / Simmons, R.L. / Kung, J. / Waight, A. / Hansen, S.K. History Deposition Feb 6, 2003 Deposition site / Processing site Revision 1.0 May 6, 2003 Provider / Type Revision 1.1 Apr 29, 2008 Group Revision 1.2 Jul 13, 2011 Group Revision 1.3 Oct 11, 2017 Group / Refinement description / Category / softwareRevision 1.4 Oct 27, 2021 Group / Database references / Derived calculationsCategory database_2 / pdbx_unobs_or_zero_occ_atoms ... database_2 / pdbx_unobs_or_zero_occ_atoms / struct_conn / struct_ref_seq_dif / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_conn.pdbx_leaving_atom_flag / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id Revision 1.5 Apr 3, 2024 Group / Refinement descriptionCategory / chem_comp_bond / pdbx_initial_refinement_modelRevision 1.6 Oct 30, 2024 Group / Category / pdbx_modification_feature
Show all Show less Remark 600 Heterogen The Br atom of the ligand was displaced during the covalent modification of the protein.
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads



















 PDBj
PDBj
















 Assembly
Assembly

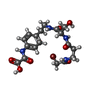
 Mass: 487.302 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H23BrN4O7
Mass: 487.302 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H23BrN4O7 Sample preparation
Sample preparation Processing
Processing