 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6weg | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of Ft (MglA-SspA)-ppGpp-PigR peptide complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSCRIPTION / Francisella tularensis / bioweapon / MglA-SspA / PigR / ppGpp | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Francisella tularensis subsp. tularensis (bacteria)Francisella tularensis (bacteria) Francisella tularensis subsp. tularensis (bacteria)Francisella tularensis (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.95 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.95 Å | ||||||
 Authors Authors | Schumacher, M.A. / Brennan, R. | ||||||
 Citation Citation | Journal: Mol Cell / Year: 2021 Title: Structural Basis for Virulence Activation of Francisella tularensis. Authors: Brady A Travis / Kathryn M Ramsey / Samantha M Prezioso / Thomas Tallo / Jamie M Wandzilak / Allen Hsu / Mario Borgnia / Alberto Bartesaghi / Simon L Dove / Richard G Brennan / Maria A Schumacher /  Abstract: The bacterium Francisella tularensis (Ft) is one of the most infectious agents known. Ft virulence is controlled by a unique combination of transcription regulators: the MglA-SspA heterodimer, PigR, ...The bacterium Francisella tularensis (Ft) is one of the most infectious agents known. Ft virulence is controlled by a unique combination of transcription regulators: the MglA-SspA heterodimer, PigR, and the stress signal, ppGpp. MglA-SspA assembles with the σ-associated RNAP holoenzyme (RNAPσ), forming a virulence-specialized polymerase. These factors activate Francisella pathogenicity island (FPI) gene expression, which is required for virulence, but the mechanism is unknown. Here we report FtRNAPσ-promoter-DNA, FtRNAPσ-(MglA-SspA)-promoter DNA, and FtRNAPσ-(MglA-SspA)-ppGpp-PigR-promoter DNA cryo-EM structures. Structural and genetic analyses show MglA-SspA facilitates σ binding to DNA to regulate virulence and virulence-enhancing genes. Our Escherichia coli RNAPσhomodimeric EcSspA structure suggests this is a general SspA-transcription regulation mechanism. Strikingly, our FtRNAPσ-(MglA-SspA)-ppGpp-PigR-DNA structure reveals ppGpp binding to MglA-SspA tethers PigR to promoters. PigR in turn recruits FtRNAP αCTDs to DNA UP elements. Thus, these studies unveil a unique mechanism for Ft pathogenesis involving a virulence-specialized RNAP that employs two (MglA-SspA)-based strategies to activate virulence genes. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6weg.cif.gz | 347.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6weg.ent.gz | 281.4 KB | Display | PDB format |
| PDBx/mmJSON format | 6weg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/we/6wegftp://data.pdbj.org/pub/pdb/validation_reports/we/6weg | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6wmpC  6wmrC  6wmtC  6wmuC  5u56S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24110.094 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Francisella tularensis subsp. tularensis (strain SCHU S4 / Schu 4) (bacteria)Strain: SCHU S4 / Schu 4 / Gene: sspA, FTT_0458 / Production host: #2: Protein | Mass: 23594.529 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Francisella tularensis (bacteria) / Gene: DR86_1530 / Production host: #3: Protein/peptide | | Mass: 2663.193 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Francisella tularensis subsp. tularensis (strain SCHU S4 / Schu 4) (bacteria)Strain: SCHU S4 / Schu 4 / Gene: FTT_0383 / Production host: #4: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION#5: Chemical | 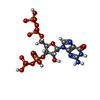  Type: RNA linking / Mass: 603.160 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N5O17P4 / Feature type: SUBJECT OF INVESTIGATION Type: RNA linking / Mass: 603.160 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N5O17P4 / Feature type: SUBJECT OF INVESTIGATIONHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.69 Å3/Da / Density % sol: 54.2 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / Details: Peg 4000, Tris HCl pH 8 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS / Beamline: 8.3.1 / Wavelength: 1.01 Å |
| Detector | Type: DECTRIS PILATUS3 S 6M / Detector: PIXEL / Date: Jul 22, 2017 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.01 Å / Relative weight: 1 |
| Reflection | Resolution: 2.95→88.461 Å / Num. obs: 22603 / % possible obs: 98.1 % / Redundancy: 2.9 % / Biso Wilson estimate: 90.75 Å2 / CC1/2: 0.997 / Rpim(I) all: 0.063 / Rsym value: 0.091 / Net I/σ(I): 7.3 |
| Reflection shell | Resolution: 2.95→3.08 Å / Num. unique obs: 2028 / CC1/2: 0.338 / Rpim(I) all: 0.95 / Rsym value: 1.38 |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5U56 Resolution: 2.95→88.461 Å / SU ML: 0.48 / Cross valid method: THROUGHOUT / σ(F): 1.35 / Phase error: 33.6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 1.13 Å / VDW probe radii: 1.2 Å / Bsol: 61.972 Å2 / ksol: 0.291 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 548.22 Å2 / Biso mean: 115.36 Å2 / Biso min: 9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.95→88.461 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -31.9861 Å / Origin y: -0.9611 Å / Origin z: -10.7611 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
