 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6un1: Crystal structure of Pseudomonas aeruginosa PBP3 in complex with ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6un1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Pseudomonas aeruginosa PBP3 in complex with temocillin | ||||||
 Components Components | Peptidoglycan D,D-transpeptidase FtsI | ||||||
 Keywords Keywords | HYDROLASE / temocillin / ticarcillin / pseudomonas aeruginosa PBP3 / penicillin-binding protein 3 / transpeptidase / HMM / high-molecular mass / b-lactam / lactam / penicillin | ||||||
| Function / homology |  Function and homology information Function and homology informationpeptidoglycan glycosyltransferase activity / serine-type D-Ala-D-Ala carboxypeptidase / serine-type D-Ala-D-Ala carboxypeptidase activity / division septum assembly / FtsZ-dependent cytokinesis / penicillin binding / peptidoglycan biosynthetic process / cell wall organization / regulation of cell shape / proteolysis / plasma membrane Similarity search - Function | ||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.26 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.26 Å | ||||||
 Authors Authors | Sacco, M. / Chen, Y. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Antimicrob.Agents Chemother. / Year: 2019 Title: Influence of the alpha-Methoxy Group on the Reaction of Temocillin with Pseudomonas aeruginosa PBP3 and CTX-M-14 beta-Lactamase. Authors: Sacco, M.D. / Kroeck, K.G. / Kemp, M.T. / Zhang, X. / Andrews, L.D. / Chen, Y. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6un1.cif.gz | 119 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6un1.ent.gz | 86.9 KB | Display | PDB format |
| PDBx/mmJSON format | 6un1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/un/6un1ftp://data.pdbj.org/pub/pdb/validation_reports/un/6un1 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6un3C  6unbC  3oc2S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 57638.719 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas aeruginosa (bacteria)Gene: pbpB, ftsI, ftsI_1, ftsI_2, ALP65_00912, CAZ10_21230, CGU42_01090, DZ934_06595, DZ962_00565, E4V10_06485, EFK68_01815, IPC1492_18840, IPC3_13380, IPC605_16140, IPC669_10550, PAERUG_E15_London_ ...Gene: pbpB, ftsI, ftsI_1, ftsI_2, ALP65_00912, CAZ10_21230, CGU42_01090, DZ934_06595, DZ962_00565, E4V10_06485, EFK68_01815, IPC1492_18840, IPC3_13380, IPC605_16140, IPC669_10550, PAERUG_E15_London_28_01_14_00534, RW109_RW109_05757 Production host: References: UniProt: Q51504, UniProt: G3XD46*PLUS, serine-type D-Ala-D-Ala carboxypeptidase |
|---|---|
| #2: Chemical | ChemComp-TJ7 / (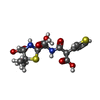  Mass: 416.469 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H20N2O7S2 / Feature type: SUBJECT OF INVESTIGATION Mass: 416.469 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H20N2O7S2 / Feature type: SUBJECT OF INVESTIGATION |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 202 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 202 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.1 Å3/Da / Density % sol: 41.3 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / Details: 20% PEG 3350 0.2 M CaOAc |
-Data collection
| Diffraction | Mean temperature: 80 K / Serial crystal experiment: N | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 19-BM / Wavelength: 1 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 210r / Detector: CCD / Date: Jun 23, 2018 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.25→50 Å / Num. obs: 22516 / % possible obs: 97.2 % / Redundancy: 4 % / Rmerge(I) obs: 0.093 / Rpim(I) all: 0.052 / Rrim(I) all: 0.107 / Χ2: 0.911 / Net I/σ(I): 6.8 / Num. measured all: 89234 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3OC2 Resolution: 2.26→33.59 Å / Cor.coef. Fo:Fc: 0.961 / Cor.coef. Fo:Fc free: 0.926 / SU B: 7.451 / SU ML: 0.178 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.358 / ESU R Free: 0.238 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 109.64 Å2 / Biso mean: 35.684 Å2 / Biso min: 19.38 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.26→33.59 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.262→2.321 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
