 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6tqi: I-MOTIF STRUCTURE FORMED FROM THE C STRAND OF A HUMAN TELOMERE FR... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6tqi | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
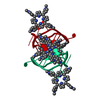
| Title | I-MOTIF STRUCTURE FORMED FROM THE C STRAND OF A HUMAN TELOMERE FRAGMENT | ||||||||||||||||||||
 Components Components | DNA (5'-* Keywords KeywordsDNA / telomeric / i-motif | Function / homology | DNA |  Function and homology information Function and homology informationBiological species |  Homo sapiens (human) Homo sapiens (human)Method |  X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.95 Å X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.95 Å  Authors AuthorsParkinson, G.N. / Wagner, A. / Viladoms-Claverol, J. / Duman, R. / El-Omari, K. |  CitationJournal: Nucleic Acids Res. / Year: 2020 CitationJournal: Nucleic Acids Res. / Year: 2020Title: Native de novo structural determinations of non-canonical nucleic acid motifs by X-ray crystallography at long wavelengths. Authors: Zhang, Y. / El Omari, K. / Duman, R. / Liu, S. / Haider, S. / Wagner, A. / Parkinson, G.N. / Wei, D. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6tqi.cif.gz | 13.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6tqi.ent.gz | 7.8 KB | Display | PDB format |
| PDBx/mmJSON format | 6tqi.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tq/6tqiftp://data.pdbj.org/pub/pdb/validation_reports/tq/6tqi | HTTPS FTP |
|---|
-Related structure data









-Links
 PDBj
PDBj

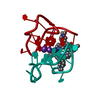
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 2683.801 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) Homo sapiens (human) |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.25 Å3/Da / Density % sol: 45.5 % / Description: hexagonal rods |
|---|---|
| Crystal grow | Temperature: 285 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: 15% PEG 400, 5 mM spermine, 50 mM 2-(N-morpholino)ethanesulfonic acid (MES) PH range: 4.5-5.5 |
-Data collection
| Diffraction |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| ||||||||||||||||||||||||
| Detector |
| ||||||||||||||||||||||||
| Radiation |
| ||||||||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||||||||
| Reflection | Resolution: 2.9→27.63 Å / Num. obs: 704 / % possible obs: 99.7 % / Redundancy: 30.1 % / CC1/2: 0.999 / Rmerge(I) obs: 0.036 / Rpim(I) all: 0.007 / Rrim(I) all: 0.037 / Net I/σ(I): 79.6 / Num. measured all: 21199 | ||||||||||||||||||||||||
| Reflection shell | Rpim(I) all: 0.011 / Diffraction-ID: 1
|

-Phasing
| Phasing | Method: SAD |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.95→25 Å / Cor.coef. Fo:Fc: 0.913 / Cor.coef. Fo:Fc free: 0.909 / SU B: 16.599 / SU ML: 0.289 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.358 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 121.8 Å2 / Biso mean: 49.393 Å2 / Biso min: 27.78 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.95→25 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.95→3.026 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
