Mass: 18.015 Da / Num. of mol.: 207 / Source method: isolated from a natural source / Formula: H2O
-
Details
Has ligand of interest
Y
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.12 Å3/Da / Density % sol: 41.92 % / Mosaicity: 0.53 °
Crystal grow
Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 0.3 M sodium chloride, 0.1 M imidazole. After growth, and before measurement, these crystals were soaked in the dye solution: saturated bromophenol blue, 0.3 M sodium chloride, 0.1 M imidazole buffer at pH 6.5.
-
Data collection
Diffraction
Mean temperature: 100 K / Serial crystal experiment: N
Diffraction source
Source: SYNCHROTRON / Site: ALBA / Beamline: XALOC / Wavelength: 0.97926 Å
Resolution: 1.38→19.39 Å / SU ML: 0.1376 / Cross valid method: FREE R-VALUE / σ(F): 1.91 / Phase error: 21.5892 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2 Details: The ligand geometry validation is not accurate because the molecule is a resonant form.
Rfactor
Num. reflection
% reflection
Rfree
0.2261
4572
5.07 %
Rwork
0.191
85624
-
obs
0.1928
90196
94.01 %
Solvent computation
Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL
Displacement parameters
Biso mean: 17.47 Å2
Refinement step
Cycle: LAST / Resolution: 1.38→19.39 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
1992
0
240
207
2439
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
X-RAY DIFFRACTION
f_bond_d
0.0072
2294
X-RAY DIFFRACTION
f_angle_d
0.8965
3142
X-RAY DIFFRACTION
f_chiral_restr
0.0666
295
X-RAY DIFFRACTION
f_plane_restr
0.0045
387
X-RAY DIFFRACTION
f_dihedral_angle_d
25.037
453
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
1.38-1.4
0.3017
130
0.2429
2817
X-RAY DIFFRACTION
90.57
1.4-1.42
0.2558
147
0.2241
2670
X-RAY DIFFRACTION
89.03
1.42-1.43
0.2572
149
0.214
2719
X-RAY DIFFRACTION
89.4
1.43-1.45
0.2589
180
0.2222
2899
X-RAY DIFFRACTION
94.71
1.45-1.47
0.2569
156
0.203
2899
X-RAY DIFFRACTION
97.29
1.47-1.49
0.2342
143
0.2003
2919
X-RAY DIFFRACTION
96.17
1.49-1.51
0.2654
133
0.1923
3001
X-RAY DIFFRACTION
96.4
1.51-1.53
0.2321
162
0.199
2888
X-RAY DIFFRACTION
95.58
1.53-1.56
0.2497
138
0.1947
2994
X-RAY DIFFRACTION
97.45
1.56-1.58
0.2301
143
0.1894
2924
X-RAY DIFFRACTION
97.09
1.58-1.61
0.2424
179
0.1886
2907
X-RAY DIFFRACTION
96.95
1.61-1.64
0.2142
157
0.1832
2986
X-RAY DIFFRACTION
96.71
1.64-1.67
0.2403
154
0.183
2773
X-RAY DIFFRACTION
93.34
1.67-1.71
0.241
136
0.1797
3023
X-RAY DIFFRACTION
96.81
1.71-1.74
0.2104
165
0.1705
2907
X-RAY DIFFRACTION
97.77
1.74-1.78
0.2168
141
0.1734
3016
X-RAY DIFFRACTION
98.41
1.78-1.83
0.2259
155
0.1768
2943
X-RAY DIFFRACTION
97.88
1.83-1.88
0.2345
162
0.1726
2983
X-RAY DIFFRACTION
97.64
1.88-1.93
0.2492
138
0.1777
2898
X-RAY DIFFRACTION
95.92
1.93-2
0.1967
182
0.1709
2917
X-RAY DIFFRACTION
95.77
2-2.06
0.2164
142
0.1722
2353
X-RAY DIFFRACTION
89.04
2.08-2.15
0.2275
123
0.1732
2188
X-RAY DIFFRACTION
92
2.15-2.25
0.2458
149
0.1791
2941
X-RAY DIFFRACTION
96.05
2.25-2.37
0.1965
160
0.1751
2949
X-RAY DIFFRACTION
97.16
2.37-2.51
0.1936
175
0.1868
2904
X-RAY DIFFRACTION
96.01
2.51-2.71
0.2202
167
0.1848
2875
X-RAY DIFFRACTION
95.75
2.71-2.98
0.2244
148
0.1911
2788
X-RAY DIFFRACTION
91.66
2.98-3.41
0.2709
143
0.187
2835
X-RAY DIFFRACTION
93.41
3.41-4.28
0.1894
152
0.1945
2896
X-RAY DIFFRACTION
95.16
4.29-19.39
0.2388
163
0.2285
2812
X-RAY DIFFRACTION
93.29
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Spain, 1items
Spain, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






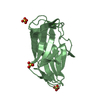
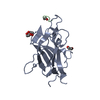




 PDBj
PDBj









 Assembly
Assembly







 Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 669.961 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C19H10Br4O5S / Feature type: SUBJECT OF INVESTIGATION
Mass: 669.961 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C19H10Br4O5S / Feature type: SUBJECT OF INVESTIGATION Mass: 69.085 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H5N2
Mass: 69.085 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H5N2 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Sample preparation
Sample preparation Processing
Processing