 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6sy1: Crystal structure of the human 2-oxoadipate dehydrogenase DHTKD1 (E1) -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6sy1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the human 2-oxoadipate dehydrogenase DHTKD1 (E1) | ||||||
 Components Components | Probable 2-oxoglutarate dehydrogenase E1 component DHKTD1, mitochondrial | ||||||
 Keywords Keywords | STRUCTURAL GENOMICS / Dehydrogenase / transketolase / thiamine pyrophosphate / Mg / Structural Genomics Consortium / SGC | ||||||
| Function / homology |  Function and homology information Function and homology informationOxidoreductases; Acting on the aldehyde or oxo group of donors; With a disulfide as acceptor / 2-oxoadipate dehydrogenase activity / OADH complex synthesizes glutaryl-CoA from 2-OA / oxoadipate dehydrogenase complex / thiamine pyrophosphate binding / hematopoietic progenitor cell differentiation / glycolytic process / generation of precursor metabolites and energy / mitochondrial matrix / mitochondrion Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.87 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.87 Å | ||||||
 Authors Authors | Bezerra, G.A. / Foster, W. / Shrestha, L. / Pena, I.A. / Coker, J. / Kolker, S. / Nicola, B.B. / von Delft, F. / Edwards, A. / Arrowsmith, C. ...Bezerra, G.A. / Foster, W. / Shrestha, L. / Pena, I.A. / Coker, J. / Kolker, S. / Nicola, B.B. / von Delft, F. / Edwards, A. / Arrowsmith, C. / Bountra, C. / Yue, W.W. / Structural Genomics Consortium (SGC) | ||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||
 Citation Citation | Journal: IUCrJ / Year: 2020 Title: Crystal structure and interaction studies of human DHTKD1 provide insight into a mitochondrial megacomplex in lysine catabolism. Authors: Gustavo A Bezerra / William R Foster / Henry J Bailey / Kevin G Hicks / Sven W Sauer / Bianca Dimitrov / Thomas J McCorvie / Jürgen G Okun / Jared Rutter / Stefan Kölker / Wyatt W Yue /   Abstract: DHTKD1 is a lesser-studied E1 enzyme among the family of 2-oxoacid de-hydrogenases. In complex with E2 (di-hydro-lipo-amide succinyltransferase, DLST) and E3 (dihydrolipo-amide de-hydrogenase, DLD) ...DHTKD1 is a lesser-studied E1 enzyme among the family of 2-oxoacid de-hydrogenases. In complex with E2 (di-hydro-lipo-amide succinyltransferase, DLST) and E3 (dihydrolipo-amide de-hydrogenase, DLD) components, DHTKD1 is involved in lysine and tryptophan catabolism by catalysing the oxidative de-carboxyl-ation of 2-oxoadipate (2OA) in mitochondria. Here, the 1.9 Å resolution crystal structure of human DHTKD1 is solved in complex with the thi-amine diphosphate co-factor. The structure reveals how the DHTKD1 active site is modelled upon the well characterized homologue 2-oxoglutarate (2OG) de-hydrogenase but engineered specifically to accommodate its preference for the longer substrate of 2OA over 2OG. A 4.7 Å resolution reconstruction of the human DLST catalytic core is also generated by single-particle electron microscopy, revealing a 24-mer cubic scaffold for assembling DHTKD1 and DLD protomers into a megacomplex. It is further demonstrated that missense DHTKD1 variants causing the inborn error of 2-amino-adipic and 2-oxoadipic aciduria impact on the complex formation, either directly by disrupting the interaction with DLST, or indirectly through destabilizing the DHTKD1 protein. This study provides the starting framework for developing DHTKD1 modulators to probe the intricate mitochondrial energy metabolism. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6sy1.cif.gz | 376.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6sy1.ent.gz | 291.9 KB | Display | PDB format |
| PDBx/mmJSON format | 6sy1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sy/6sy1ftp://data.pdbj.org/pub/pdb/validation_reports/sy/6sy1 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2jgdS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 100926.102 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: DHTKD1, KIAA1630 / Production host:  References: UniProt: Q96HY7, oxoglutarate dehydrogenase (succinyl-transferring) #2: Chemical | 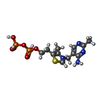  Mass: 425.314 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N4O7P2S Mass: 425.314 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N4O7P2S#3: Chemical |   Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1066 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1066 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.32 Å3/Da / Density % sol: 47.09 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion / pH: 8.5 Details: 20% PEG3350 -- 10% ethylene glycol -- 0.1M bis-tris-propane pH 8.5 -- 0.2M sodium formate |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond / Beamline: I03 / Wavelength: 0.9762 Å | ||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS3 6M / Detector: PIXEL / Date: Nov 25, 2018 | ||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9762 Å / Relative weight: 1 | ||||||||||||||||||||||||
| Reflection | Resolution: 1.87→46.01 Å / Num. obs: 145107 / % possible obs: 96.6 % / Redundancy: 1.8 % / Biso Wilson estimate: 16.081 Å2 / Rpim(I) all: 0.102 / Rrim(I) all: 0.144 / Net I/σ(I): 5 / Num. measured all: 254605 | ||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2JGD Resolution: 1.87→46.006 Å / SU ML: 0.23 / Cross valid method: THROUGHOUT / σ(F): 1.96 / Phase error: 25.29
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 92.18 Å2 / Biso mean: 26.2993 Å2 / Biso min: 10.71 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.87→46.006 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0
|
