 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 6sii | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | T. brucei FPPS in complex with 1-((1H-indol-3-yl)methyl)-N-(3-chlorobenzyl)piperidin-4-amine | |||||||||
 要素 要素 | Farnesyl pyrophosphate synthase | |||||||||
 キーワード キーワード | TRANSFERASE / sterol biosynthesis / homodimer / farnesyl pyrophosphate synthase / Trypanosoma brucei | |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報trans, trans-farnesyl diphosphate biosynthetic process / dimethylallyltranstransferase activity / (2E,6E)-farnesyl diphosphate synthase activity / metal ion binding / cytoplasm 類似検索 - 分子機能 | |||||||||
| 生物種 |  | |||||||||
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 2.33 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 2.33 Å | |||||||||
 データ登録者 データ登録者 | Muenzker, L. / Petrick, J.K. / Schleberger, C. / Jahnke, W. | |||||||||
| 資金援助 |  スイス, 2件 スイス, 2件
| |||||||||
 引用 引用 | ジャーナル: To Be Published タイトル: T. brucei FPPS 著者: Muenzker, L. / Jahnke, W. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 6sii.cif.gz | 151 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb6sii.ent.gz | 119.1 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 6sii.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/si/6siiftp://data.pdbj.org/pub/pdb/validation_reports/si/6sii | HTTPS FTP |
|---|
-関連構造データ
| 関連構造データ |  4rypS S: 精密化の開始モデル |
|---|---|
| 類似構造データ |










-リンク
 PDBj
PDBj
- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 単位格子 |
|
-要素
| #1: タンパク質 | 分子量: 42169.211 Da / 分子数: 1 / 由来タイプ: 組換発現 / 詳細: GP is an expression tag 由来: (組換発現) 発現宿主:  |
|---|---|
| #2: 化合物 | ChemComp-LEZ / ~{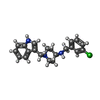  分子量: 353.888 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C21H24ClN3 / タイプ: SUBJECT OF INVESTIGATION 分子量: 353.888 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C21H24ClN3 / タイプ: SUBJECT OF INVESTIGATION |
| #3: 化合物 | ChemComp-GOL /   分子量: 92.094 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C3H8O3 分子量: 92.094 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C3H8O3 |
| 研究の焦点であるリガンドがあるか | Y |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.22 Å3/Da / 溶媒含有率: 44.71 % / 解説: needles |
|---|---|
| 結晶化 | 温度: 293 K / 手法: 蒸気拡散法, シッティングドロップ法 / 詳細: 0.12 M CsCl 12 % w/v PEG 3350, 12 % v/v DMSO |
-データ収集
| 回折 | 平均測定温度: 100 K / Serial crystal experiment: N |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: SLS / ビームライン: X10SA / 波長: 1.00002 Å |
| 検出器 | タイプ: DECTRIS PILATUS3 6M / 検出器: PIXEL / 日付: 2018年8月27日 |
| 放射 | プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.00002 Å / 相対比: 1 |
| 反射 | 解像度: 2.326→57.041 Å / Num. obs: 17624 / % possible obs: 99.4 % / 冗長度: 18.5 % / Biso Wilson estimate: 79.33 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.113 / Rpim(I) all: 0.027 / Rrim(I) all: 0.116 / Rsym value: 0.113 / Net I/σ(I): 15.1 / Num. measured all: 326497 |
| 反射 シェル | 解像度: 2.326→2.366 Å / 冗長度: 19.6 % / Rmerge(I) obs: 12.631 / Mean I/σ(I) obs: 0.3 / Num. measured all: 48808 / Num. unique obs: 2512 / CC1/2: 0.59 / Rpim(I) all: 1.983 / Rrim(I) all: 8.823 / Rsym value: 12.631 / Net I/σ(I) obs: 0.4 / % possible all: 100 |
-位相決定
| 位相決定 | 手法: 分子置換 | ||||||
|---|---|---|---|---|---|---|---|
| Phasing MR | Model details: Phaser MODE: MR_TRA
|
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 分子置換 開始モデル: pdbid 4ryp 解像度: 2.33→57.04 Å / Cor.coef. Fo:Fc: 0.944 / Cor.coef. Fo:Fc free: 0.926 / SU R Cruickshank DPI: 0.394 / 交差検証法: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.429 / SU Rfree Blow DPI: 0.266 / SU Rfree Cruickshank DPI: 0.261
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso max: 228.35 Å2 / Biso mean: 121.49 Å2 / Biso min: 64.15 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.49 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: final / 解像度: 2.33→57.04 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 2.33→2.38 Å / Rfactor Rfree error: 0 / Total num. of bins used: 41
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 TLS | 手法: refined / Origin x: 3.4606 Å / Origin y: 11.916 Å / Origin z: -11.451 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 TLSグループ | Selection details: { A|* } |
