 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
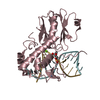
Yorodumi- PDB-6rnm: Crystal structure of a complex between the LlFpg protein, a THF-D... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6rnm | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of a complex between the LlFpg protein, a THF-DNA and an inhibitor | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE / DNA glycosylase complex / inhibitor | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA-formamidopyrimidine glycosylase / oxidized purine nucleobase lesion DNA N-glycosylase activity / 8-oxo-7,8-dihydroguanine DNA N-glycosylase activity / class I DNA-(apurinic or apyrimidinic site) endonuclease activity / DNA-(apurinic or apyrimidinic site) lyase / nucleotide-excision repair / base-excision repair / damaged DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |  Lactococcus lactis subsp. cremoris (lactic acid bacteria) Lactococcus lactis subsp. cremoris (lactic acid bacteria)synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.76 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.76 Å | ||||||
 Authors Authors | Coste, F. / Goffinont, S. / Castaing, B. | ||||||
 Citation Citation | Journal: Int J Mol Sci / Year: 2020 Title: Thiopurine Derivative-Induced Fpg/Nei DNA Glycosylase Inhibition: Structural, Dynamic and Functional Insights. Authors: Rieux, C. / Goffinont, S. / Coste, F. / Tber, Z. / Cros, J. / Roy, V. / Guerin, M. / Gaudon, V. / Bourg, S. / Biela, A. / Aucagne, V. / Agrofoglio, L. / Garnier, N. / Castaing, B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6rnm.cif.gz | 167.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6rnm.ent.gz | 128 KB | Display | PDB format |
| PDBx/mmJSON format | 6rnm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 6rnm_validation.pdf.gz | 722.4 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 6rnm_full_validation.pdf.gz | 722.4 KB | Display | |
| Data in XML | 6rnm_validation.xml.gz | 17.7 KB | Display | |
| Data in CIF | 6rnm_validation.cif.gz | 27.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rn/6rnmftp://data.pdbj.org/pub/pdb/validation_reports/rn/6rnm | HTTPS FTP |
-Related structure data
| Related structure data |  6rnoC  6rnrC  6ro2C  6rokC  6rp0C  6rp7C  1pm5S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj







































- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 31116.217 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Lactococcus lactis subsp. cremoris (lactic acid bacteria)Gene: mutM, fpg, NCDO763_0992 / Production host: References: UniProt: A0A165FVI1, UniProt: P42371*PLUS, DNA-formamidopyrimidine glycosylase, DNA-(apurinic or apyrimidinic site) lyase |
|---|
-DNA chain , 2 types, 2 molecules DE
| #2: DNA chain | Mass: 4054.614 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) |
|---|---|
| #3: DNA chain | Mass: 4355.884 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) |
-Non-polymers , 4 types, 453 molecules 


| #4: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn | ||
|---|---|---|---|
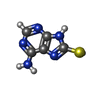
| #5: Chemical | ChemComp-KB5 /  Mass: 167.192 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H5N5S / Feature type: SUBJECT OF INVESTIGATION Mass: 167.192 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H5N5S / Feature type: SUBJECT OF INVESTIGATION | ||
| #6: Chemical |  Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 449 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.8 Å3/Da / Density % sol: 67.65 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / Details: HEPES, SODIUM CITRATE |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: MASSIF-3 / Wavelength: 0.9677 Å / Beamline: MASSIF-3 / Wavelength: 0.9677 Å |
| Detector | Type: DECTRIS EIGER X 4M / Detector: PIXEL / Date: Jul 11, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9677 Å / Relative weight: 1 |
| Reflection | Resolution: 1.76→59.13 Å / Num. obs: 61222 / % possible obs: 99.9 % / Redundancy: 7.2 % / Biso Wilson estimate: 32.67 Å2 / Rmerge(I) obs: 0.056 / Net I/σ(I): 18.3 |
| Reflection shell | Resolution: 1.76→1.79 Å / Redundancy: 7.5 % / Rmerge(I) obs: 0.957 / Num. unique obs: 3017 / % possible all: 99.3 |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1PM5 Resolution: 1.76→59.13 Å / Cor.coef. Fo:Fc: 0.967 / Cor.coef. Fo:Fc free: 0.96 / Rfactor Rfree error: 0 / SU R Cruickshank DPI: 0.077 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.085 / SU Rfree Blow DPI: 0.082 / SU Rfree Cruickshank DPI: 0.076
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 112.71 Å2 / Biso mean: 38.53 Å2 / Biso min: 18.12 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.21 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.76→59.13 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.76→1.81 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
